
Label-Free Quantitative Analysis of Pig Liver Proteome after Hepatitis E Virus Infection

 , , , , , ,
, , , , , ,
Abstract
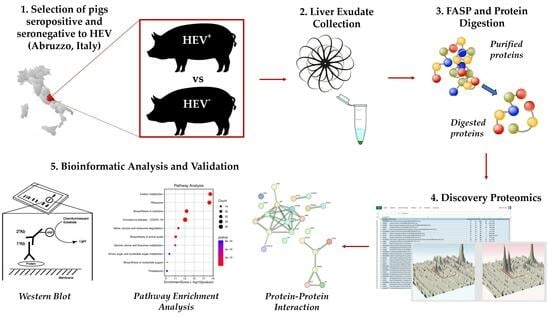
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Experimental Plan
2.2. Enzyme-Linked Immunosorbent Assay (ELISA) for the Identification of HEV Antibodies in Blood Serum
2.3. Liver Exudate Collection and Filter-Aided Sample Preparation (FASP)
2.4. Liquid Chromatography–Mass Spectrometry (LC-MS/MS) and Label-Free Quantitative Profiling
2.5. Bioinformatics for Functional and Protein Network Analyses
2.6. Western Blot Analysis
3. Results
3.1. Selection of Animals That Suffered the HEV Infection
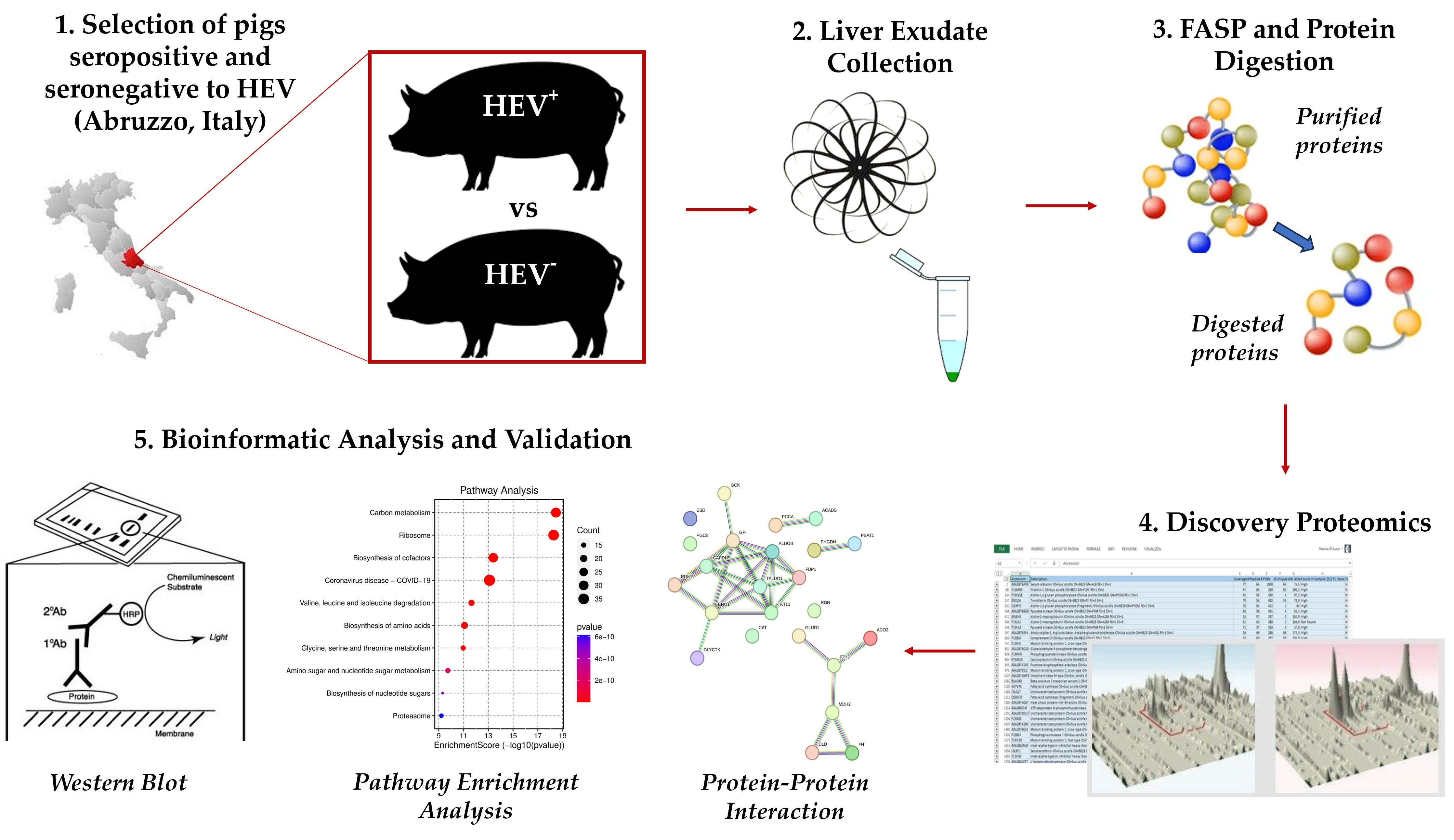
3.2. Protein Characterization in Pig Liver
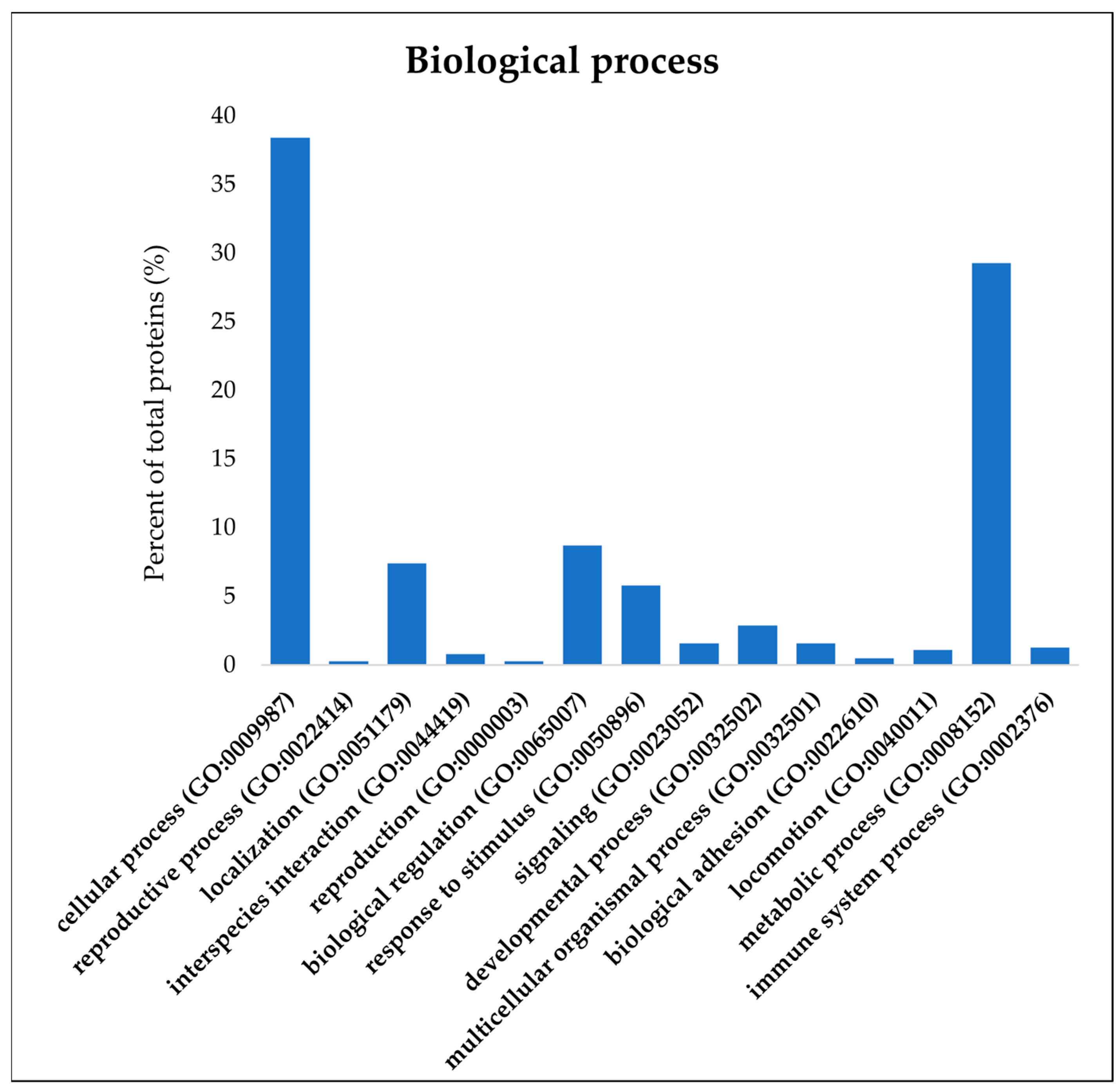
3.3. Comparative Proteomic Analysis of Pig Liver Exudate Negative and Positive to HEV Infection
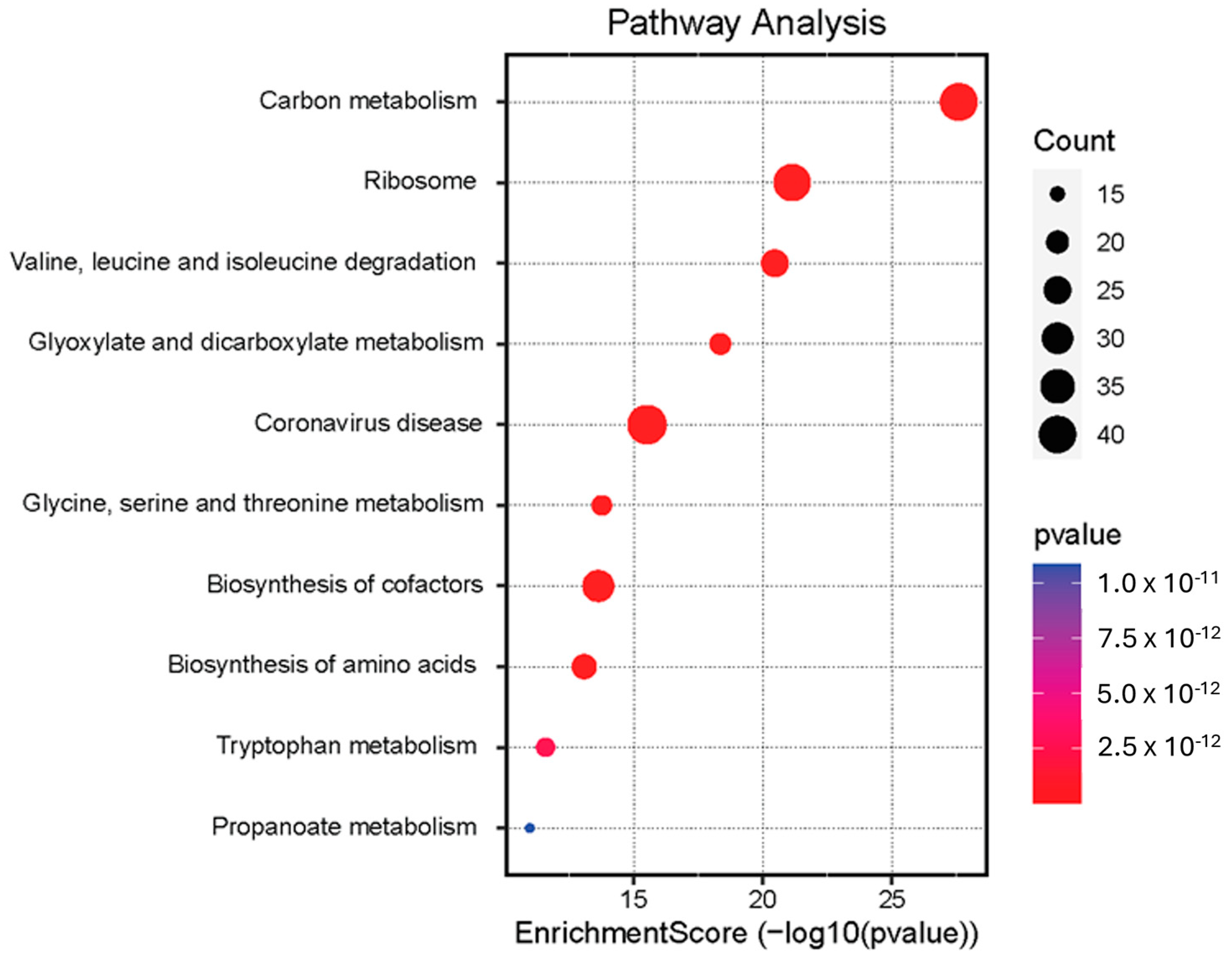
3.4. Pathway Enrichment Analysis
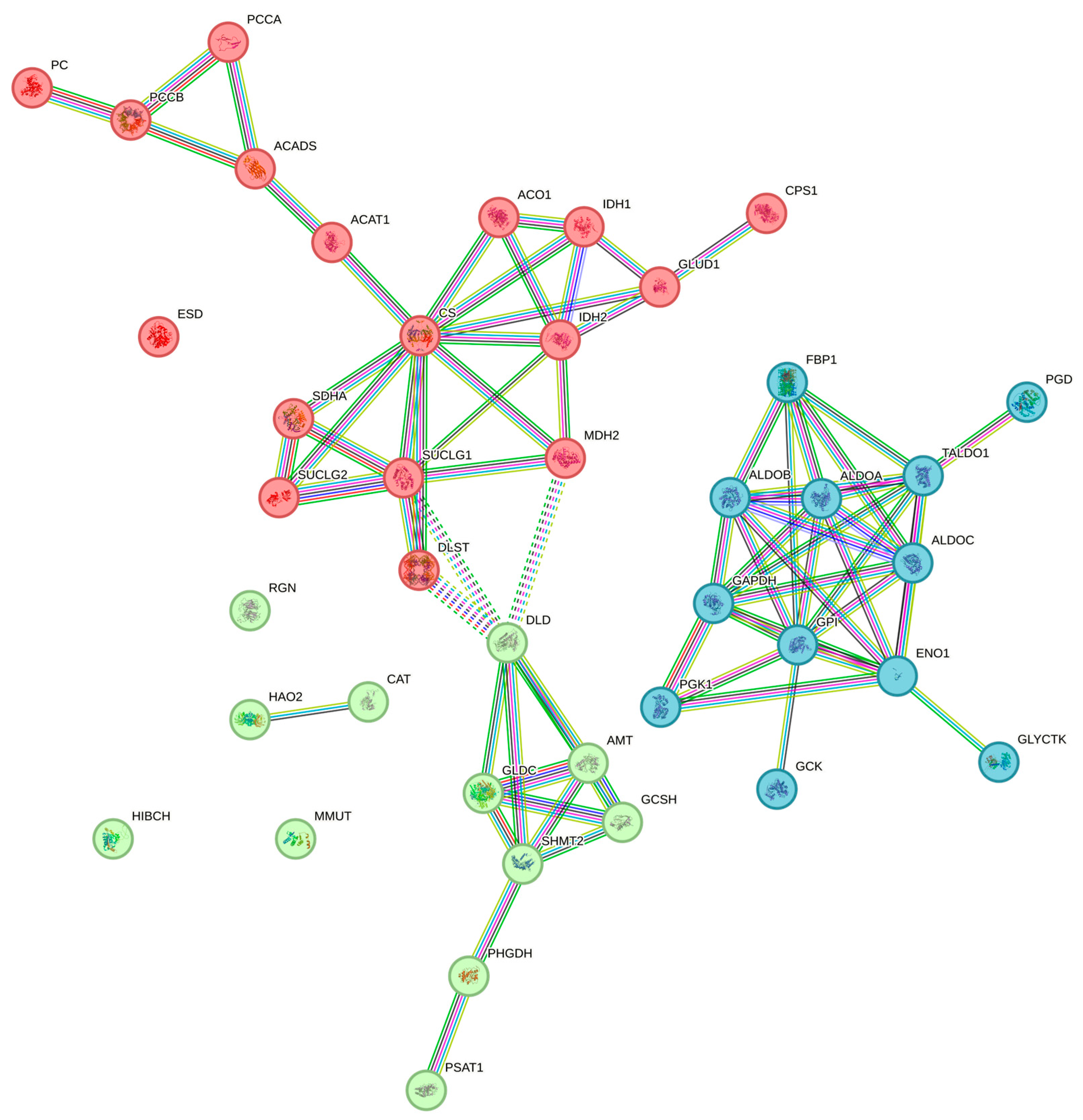
3.5. Evaluation of Protein–Protein Interaction (PPI)
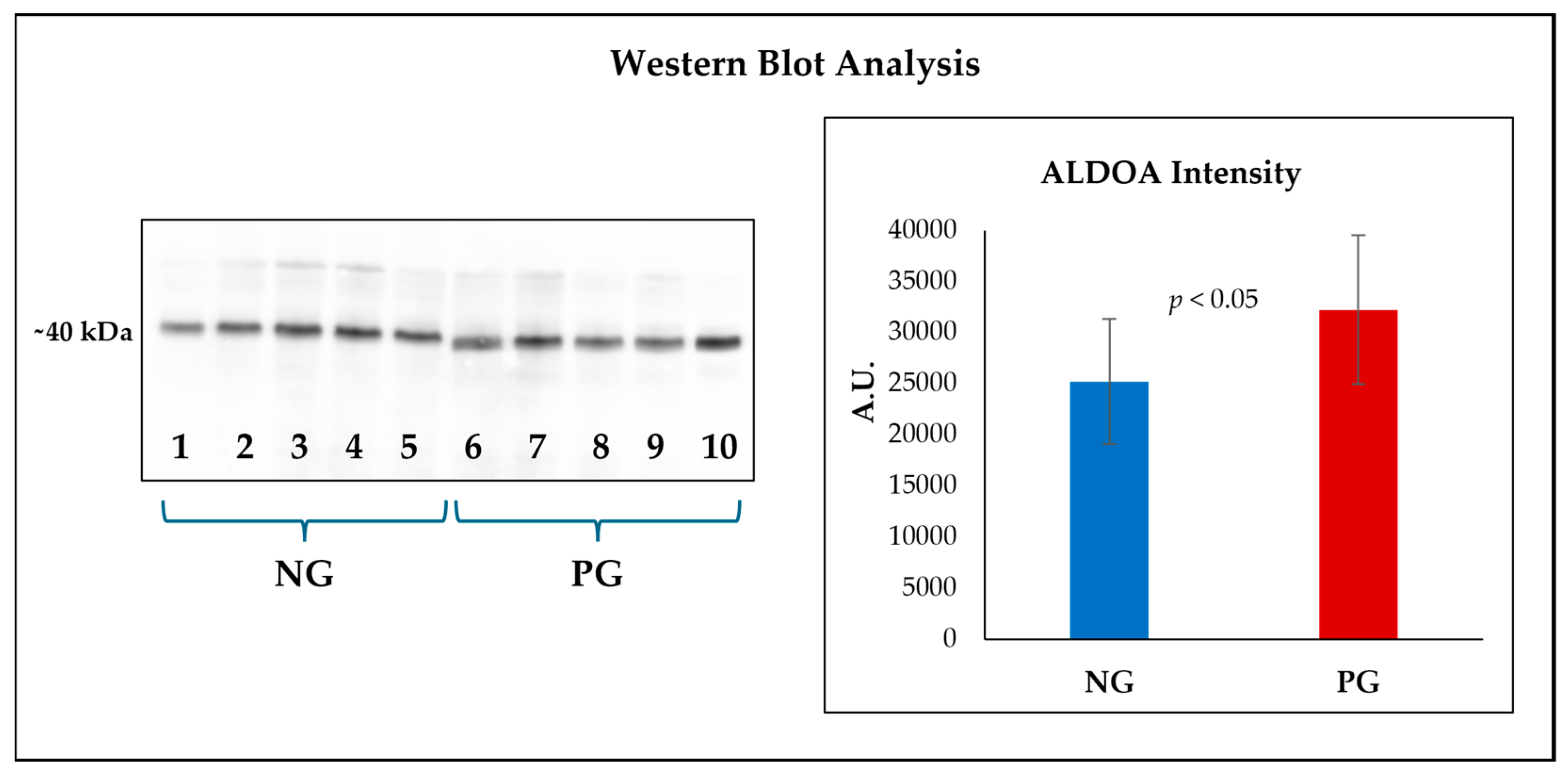
3.6. Confirmation of the Differential Expression of Fructose-Bisphosphate Aldolase a by Western Blot
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, P.; Liu, J.; Li, Y.; Su, J.; Ma, Z.; Bramer, W.M.; Cao, W.; De Man, R.A.; Peppelenbosch, M.P.; Pan, Q. The global epidemiology of hepatitis E virus infection: A systematic review and meta-analysis. Liver Int. 2020, 40, 1516–1528. [Google Scholar] [CrossRef]
- Graff, J.; Torian, U.; Nguyen, H.; Emerson, S.U. A bicistronic subgenomic mRNA encodes both the ORF2 and ORF3 proteins of hepatitis E virus. J. Virol. 2006, 80, 5919–5926. [Google Scholar] [CrossRef]
- Smith, D.B.; Simmonds, P.; Jameel, S.; Emerson, S.U.; Harrison, T.J.; Meng, X.-J.; Okamoto, H.; Van Der Poel, W.H.M.; Purdy, M.A.; International Committee on the Taxonomy of Viruses Hepeviridae Study Group. Consensus proposals for classification of the family Hepeviridae. J. Gen. Virol. 2014, 95, 2223–2232. [Google Scholar] [CrossRef]
- Thakur, V.; Ratho, R.K.; Kumar, S.; Saxena, S.K.; Bora, I.; Thakur, P. Viral hepatitis E and chronicity: A growing public health concern. Front. Microbiol. 2020, 11, 577339. [Google Scholar] [CrossRef]
- Kamani, L.; Padhani, Z.A.; Das, J.K. Hepatitis E: Genotypes, strategies to prevent and manage, and the existing knowledge gaps. JGH Open 2021, 5, 1127–1134. [Google Scholar] [CrossRef] [PubMed]
- Izopet, J.; Tremeaux, P.; Marion, O.; Migueres, M.; Capelli, N.; Chapuy-Regaud, S.; Mansuy, J.-M.; Abravanel, F.; Kamar, N.; Lhomme, S. Hepatitis E virus infections in Europe. J. Clin. Virol. 2019, 120, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Ricci, A.; Allende, A.; Bolton, D.; Chemaly, M.; Davies, R.; Escamez, P.S.F.; Herman, L.; Koutsoumanis, K.; Lindqvist, R.; Norrung, B.; et al. Public health risks associated with hepatitis E virus (HEV) as a food-borne pathogen. EFSA J. 2017, 15, e04886. [Google Scholar] [CrossRef]
- Andraud, M.; Casas, M.; Pavio, N.; Rose, N. Early-Life Hepatitis E Infection in Pigs: The Importance of Maternally-Derived Antibodies. PLoS ONE 2014, 9, e105527. [Google Scholar] [CrossRef] [PubMed]
- Meester, M.; Tobias, T.J.; Bouwknegt, M.; Kusters, N.E.; Stegeman, J.A.; Van der Poel, W.H. Infection dynamics and persistence of hepatitis E virus on pig farms—A review. Porcine Health Manag. 2021, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.Y.; Qian, W.J.; Diamond, D.L.; Liu, T.; Gritsenko, M.A.; Monroe, M.E.; Camp II, D.G.; Smith, R.D.; Katze, M.G. Quantitative analysis of human immunodeficiency virus type 1-infected CD4+ cell proteome: Dysregulated cell cycle progression and nuclear transport coincide with robust virus production. J. Virol. 2007, 81, 7571–7583. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.S.; Tang, L.Y.; Dai, J.; Zhou, H.; Li, S.J.; Xia, Q.C.; Wu, J.R.; Zeng, R. Quantitative Analysis of Severe Acute Respiratory Syndrome (SARS)-associated Coronavirus-infected Cells Using Proteomic Approaches: Implications for Cellular Responses to Virus Infection. Mol. Cell. Proteom. 2005, 4, 902–913. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Han, D.; Song, J.Y.; Kim, J.H.; Yoon, S. Proteomic analysis of swine hepatitis E virus (sHEV)-infected livers reveals upregulation of apolipoprotein and downregulation of ferritin heavy chain. FEMS Immunol. Med. Microbiol. 2001, 61, 359–363. [Google Scholar] [CrossRef]
- Rogée, S.; Le Gall, M.; Chafey, P.; Bouquet, J.; Cordonnier, N.; Frederici, C.; Pavio, N. Quantitative proteomics identifies host factors modulated during acute hepatitis E virus infection in the swine model. J. Virol. 2015, 89, 129–143. [Google Scholar] [CrossRef]
- Council Regulation (RC). Consolidated Text: Council Regulation (EC) No 1099/2009 of 24 September 2009 on the Protection of Animals at the Time of Killing; European Union: Brussels, Belgium, 2009. [Google Scholar]
- European Union. Directive 2010/63/EU of the European Parliament and of the Council of 22 September 2010 on the Protection of Animals Used for Scientific Purposes. 2010. Available online: http://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32010L0063&from=EN (accessed on 11 January 2022).
- Legislative Decree 26/2014 of 4 March 2014 Implementing Directive 2010/63/EU on the Protection of Animals Used for Scientific Purposes. Available online: https://www.gazzettaufficiale.it/eli/id/2014/03/14/14G00036/sg (accessed on 28 February 2022).
- Di Luca, A.; Ianni, A.; Bennato, F.; Martino, C.; Henry, M.; Meleady, P.; Martino, G. Comparative Proteomics Analysis of Pig Muscle Exudate through Label-Free Liquid Chromatography-Mass Spectrometry. Animals 2023, 13, 1460. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248. [Google Scholar] [CrossRef]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Meth. 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Coleman, O.; Henry, M.; Clynes, M.; Meleady, P. Filter-Aided Sample Preparation (FASP) for Improved Proteome Analysis of Recombinant Chinese Hamster Ovary Cells. Methods Mol. Biol. 2017, 1603, 187–194. [Google Scholar] [CrossRef]
- Di Luca, A.; Henry, M.; Meleady, P.; O’connor, R. Label-free LC-MS analysis of HER2+ breast cancer cell line response to HER2 inhibitor treatment. DARU J. Pharm. Sci. 2015, 23, 40. [Google Scholar] [CrossRef]
- Thomas, P.D.; Ebert, D.; Muruganujan, A.; Mushayahama, T.; Albou, L.P.; Mi, H. PANTHER: Making genome-scale phylogenetics accessible to all. Protein Sci. 2022, 31, 8–22. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Mauceri, C.; Clemente, M.G.; Castiglia, P.; Antonucci, R.; Schwarz, K.B. Hepatitis E in Italy: A silent presence. J. Infect. Public Health 2018, 11, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Picchi, G.; Ricciardi, A.; Marola, L.; Di Norcia, M.; D’alessandro, M.; Mariani, R.; Cacciatore, P.; Sozio, F.; Necozione, S.; Cofini, V.; et al. Prevalence of HEV infection in acute non-ABC hepatitis and prognostic role of extrahepatic manifestations. Infez. Med. 2022, 31, 70. [Google Scholar] [CrossRef] [PubMed]
- Martino, C.; Rampacci, E.; Pierini, I.; Giammarioli, M.; Stefanetti, V.; Hyatt, D.R.; Ianni, A.; Di Paolo, G.; Coletti, M.; Passamonti, F. Detection of anti-HEV antibodies and RNA of HEV in pigs from a hyperendemic Italian region with high human seroprevalence. Eur. J. Public Health 2021, 31, 68–72. [Google Scholar] [CrossRef]
- Shen, Q.; Pu, Y.; Fu, X.; Xie, Y.; Bian, X.; Yang, S.; Yang, Y.; Cui, L.; Wang, X.; Wang, H.; et al. Changes in the cellular proteins of A549 infected with Hepatitis E virus by proteomics analysis. BMC Vet. Res. 2014, 10, 188. [Google Scholar] [CrossRef]
- González, R.; Butković, A.; Elena, S.F. From foes to friends: Viral infections expand the limits of host phenotypic plasticity. Adv. Virus Res. 2020, 106, 85–121. [Google Scholar] [CrossRef]
- Aubin-Horth, N.; Renn, S.C. Genomic reaction norms: Using integrative biology to understand molecular mechanisms of phenotypic plasticity. Mol. Ecol. 2009, 18, 3763–3780. [Google Scholar] [CrossRef]
- Chandel, N.S. Glycolysis. Cold Spring Harbor Perspect. Biol. 2021, 13, a040535. [Google Scholar] [CrossRef]
- Moin, S.M.; Chandra, V.; Arya, R.; Jameel, S. The hepatitis E virus ORF3 protein stabilizes HIF-1α and enhances HIF-1-mediated transcriptional activity through p300/CBP. Cell. Microbiol. 2009, 11, 1409–1421. [Google Scholar] [CrossRef] [PubMed]
- Korkaya, H.; Jameel, S.; Gupta, D.; Tyagi, S.; Kumar, R.; Zafrullah, M.; Mazumdar, M.; Lal, S.K.; Xiaofang, L.; Sehgal, D.; et al. The ORF3 protein of hepatitis E virus binds to Src homology 3 domains and activates MAPK. J. Biol. Chem. 2001, 276, 42389–42400. [Google Scholar] [CrossRef] [PubMed]
- Emerson, S.U.; Nguyen, H.; Torian, U.; Purcell, R.H. ORF3 protein of hepatitis E virus is not required for replication, virion assembly, or infection of hepatoma cells in vitro. J. Virol. 2006, 80, 10457–10464. [Google Scholar] [CrossRef]
- Lonergan, S.M.; Stalder, K.J.; Huff-Lonergan, E.; Knight, T.J.; Goodwin, R.N.; Prusa, K.J.; Beitz, D.C. Influence of lipid content on pork sensory quality within pH classification. J. Anim. Sci. 2007, 85, 1074–1079. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, D.N.; Miller, K.D.; Ellis, M.; McKeith, F.K.; Wilson, E.R. Relationships between longissimus glycolytic potential and swine growth performance, carcass traits, and pork quality. J. Anim. Sci. 2003, 81, 2206–2212. [Google Scholar] [CrossRef] [PubMed]
- Citores, L.; Iglesias, R.; Ferreras, J.M. Antiviral activity of ribosome-inactivating proteins. Toxins 2021, 13, 80. [Google Scholar] [CrossRef]
- Ahmad, I.; Holla, R.P.; Jameel, S. Molecular virology of hepatitis E virus. Virus Res. 2011, 161, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Ratra, R.; Kar-Roy, A.; Lal, S.K. ORF3 protein of hepatitis E virus interacts with the B β chain of fibrinogen resulting in decreased fibrinogen secretion from HuH-7 cells. J. Gen. Virol. 2009, 90, 1359–1370. [Google Scholar] [CrossRef]
- Cieśla, M.; Mierzejewska, J.; Adamczyk, M.; Farrants, A.K.Ö.; Boguta, M. Fructose bisphosphate aldolase is involved in the control of RNA polymerase III-directed transcription. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 1103–1110. [Google Scholar] [CrossRef]
- Geiduschek, E.P.; Kassavetis, G.A. The RNA polymerase III transcription apparatus11Edited by PE Wright. J. Mol. Biol. 2001, 310, 1–26. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UniProt ID 1 | Gene Name | Protein Description | Peptide 2 | Score 3 | Anova (p) | Fold Change |
|---|---|---|---|---|---|---|
| A0A481B0J7 | CPS1 | carbamoyl-phosphate synthase [ammonia], mitochondrial | 78 | 457.27 | 0.0049 | 2.04 |
| A0A8D1CLI4 | FLNA | filamin-A isoform X2 | 27 | 99.35 | 0.0010 | 2.12 |
| A0A480SKT5 | DMGDH | dimethylglycine dehydrogenase, mitochondrial isoform X1 | 25 | 125.00 | 0.0061 | 2.75 |
| A0A287ATN8 | HSPD1 | 60 kDa heat shock protein, mitochondrial | 24 | 147.20 | 0.0039 | 1.98 |
| P42174 | GLUD1 | glutamate dehydrogenase 1, mitochondrial | 23 | 133.50 | 0.0007 | 2.01 |
| Q7YS28 | PC | pyruvate carboxylase, mitochondrial | 23 | 113.27 | 0.0033 | 10.54 |
| A0A4X1URZ5 | ACSS3 | acyl-CoA synthetase medium-chain family member 4 | 22 | 121.18 | 0.0017 | 2.01 |
| A0A480SA59 | SARDH | sarcosine dehydrogenase, mitochondrial isoform X1 | 22 | 107.97 | 0.0004 | 2.96 |
| A0A287ADJ2 | HSPA9 | stress-70 protein, mitochondrial | 20 | 84.28 | 0.0001 | 2.50 |
| Q2XQV4 | ALDH2 | aldehyde dehydrogenase, mitochondrial precursor | 18 | 94.69 | 0.0026 | 1.78 |
| A0A4X1UM84 | PCK1 | phosphoenolpyruvate carboxykinase, cytosolic [GTP] | 18 | 98.33 | 0.0036 | 2.90 |
| P26234 | VCL | Vinculin | 18 | 63.18 | 0.0007 | 2.28 |
| P0DTA4 | PCCA | propionyl-CoA carboxylase alpha chain, mitochondrial | 18 | 78.68 | 0.0025 | 1.98 |
| A0A8D1MNH9 | SPTAN1 | spectrin alpha chain, non-erythrocytic 1 | 17 | 65.06 | 0.0000 | 2.68 |
| P33198 | IDH2 | isocitrate dehydrogenase, mitochondrial | 16 | 66.34 | 0.0113 | 1.53 |
| P79384 | PCCB | propionyl-CoA carboxylase beta chain, mitochondrial precursor | 16 | 70.50 | 0.0173 | 1.63 |
| A0A8D1EFW2 | AGXT2 | alanine-glyoxylate aminotransferase 2, mitochondrial | 16 | 87.66 | 0.0012 | 1.98 |
| D0G0B3 | ACAA2 | 3-ketoacyl-CoA thiolase, mitochondrial | 16 | 99.47 | 0.0057 | 2.21 |
| A0A4X1UM84 | PCK1 | phosphoenolpyruvate carboxykinase, cytosolic [GTP] | 16 | 94.81 | 0.0055 | 1.90 |
| A0A287BBX3 | NADK2 | NAD kinase 2, mitochondrial | 16 | 76.07 | 0.0016 | 1.98 |
| O19072 | OTC | ornithine transcarbamylase, mitochondrial | 15 | 94.18 | 0.0012 | 2.04 |
| P50441 | GATM | glycine amidinotransferase, mitochondrial | 15 | 92.88 | 0.0003 | 2.22 |
| A0A4X1V4W9 | HSDL2 | hydroxysteroid dehydrogenase-like protein 2 | 15 | 68.16 | 0.0003 | 2.69 |
| I3LJ48 | EHHADH | peroxisomal bifunctional enzyme | 14 | 61.42 | 0.0007 | 2.01 |
| A0A8D1GE69 | ACADSB | short/branched chain specific acyl-CoA dehydrogenase, mitochondrial | 14 | 80.79 | 0.0102 | 2.04 |
| A0A8D0XUW7 | ATP5F1B | ATP synthase subunit beta, mitochondrial | 14 | 65.54 | 0.0032 | 3.05 |
| A0A287AIP7 | ACSL1 | long-chain-fatty-acid--CoA ligase 1 | 13 | 48.59 | 0.0061 | 1.73 |
| A0A4X1URZ5 | ACSS3 | acyl-CoA synthetase short-chain family member 3, mitochondrial | 13 | 55.80 | 0.0003 | 3.38 |
| P80229 | SERPINB1 | leukocyte elastase inhibitor | 12 | 48.80 | 0.0026 | 3.98 |
| A0A287BLE0 | FLNB | filamin-B | 12 | 44.89 | 0.0023 | 2.03 |
| C0MHR2 | CLTC | clathrin heavy chain | 12 | 45.85 | 0.0218 | 2.28 |
| A0A8D1UET6 | TST | thiosulfate sulfurtransferase | 12 | 75.14 | 0.0022 | 1.89 |
| A0A286ZND5 | PRDX1 | peroxiredoxin-1 | 12 | 60.03 | 0.0087 | 2.88 |
| P80021 | ATP5F1A | ATP synthase subunit alpha, mitochondrial | 12 | 65.71 | 0.0066 | 2.73 |
| Q29554 | HADHA | trifunctional enzyme subunit alpha, mitochondrial | 11 | 44.65 | 0.0039 | 4.12 |
| F1RMF7 | GLYAT | glycine N-acyltransferase | 11 | 56.90 | 0.0150 | 1.58 |
| I3LP02 | ACAT1 | acetyl-CoA acetyltransferase, mitochondrial | 11 | 54.70 | 0.0119 | 1.87 |
| Q6UAQ8 | ETFB | electron transfer flavoprotein subunit beta | 11 | 54.83 | 0.0067 | 2.04 |
| Q9XT00 | HSD17B8 | 3-hydroxyacyl-CoA dehydrogenase | 11 | 85.09 | 0.0000 | 2.42 |
| F2Z5N5 | RPL26L1 | ceruloplasmin isoform X1 | 10 | 59.12 | 0.0027 | 2.46 |
| F1SAD9 | PDIA4 | protein disulfide-isomerase A4 | 10 | 36.38 | 0.0269 | 2.21 |
| P53590 | SUCLG2 | succinate-CoA ligase [GDP-forming] subunit beta, mitochondrial | 10 | 58.96 | 0.0036 | 2.32 |
| B2ZF49 | HADHA | enoyl-CoA hydratase, mitochondrial | 10 | 48.23 | 0.0102 | 1.89 |
| P00346 | MDH2 | malate dehydrogenase, mitochondrial | 10 | 63.15 | 0.0006 | 1.89 |
| A0A4X1UTH9 | IVD | isovaleryl-CoA dehydrogenase, mitochondrial | 10 | 62.96 | 0.0032 | 2.02 |
| P41367 | ACADM | medium-chain specific acyl-CoA dehydrogenase, mitochondrial | 10 | 47.06 | 0.0003 | 2.85 |
| A0A8W4F721 | ALDH4A1 | delta-1-pyrroline-5-carboxylate dehydrogenase, mitochondrial | 10 | 42.48 | 0.0035 | 1.93 |
| UniProt ID 1 | Gene Name | Protein Description | Peptide 2 | Score 3 | Anova (p) | Fold Change |
|---|---|---|---|---|---|---|
| Q58GK8 | FASN | fatty acid synthase | 24 | 124.48 | 0.0194 | 1.82 |
| Q95332 | BHMT | betaine-homocysteine S-methyltransferase 1 | 19 | 140.67 | 0.0003 | 1.78 |
| L7TEV7 | AOX1 | aldehyde oxidase | 18 | 91.88 | 0.0072 | 2.06 |
| F1S1E7 | DPYS | dihydropyrimidinase isoform X1 | 17 | 108.90 | 0.0015 | 1.86 |
| A0A4X1VFQ7 | LOC110255172 | acyl-coenzyme A amino acid N-acyltransferase 2-like | 17 | 93.37 | 0.0078 | 1.75 |
| A0A480ST43 | ASS1 | argininosuccinate synthase | 16 | 97.76 | 0.0015 | 1.66 |
| F1S3H8 | RDH8 | retinol dehydrogenase 1 | 16 | 76.58 | 0.0276 | 1.54 |
| A0A4X1UZ96 | UPB1 | beta-ureidopropionase | 16 | 94.43 | 0.0086 | 1.98 |
| P23687 | PREP | prolyl endopeptidase | 16 | 71.89 | 0.0139 | 2.11 |
| Q28943 | DPYD | dihydropyrimidine dehydrogenase [NADP(+)] precursor | 15 | 67.95 | 0.0017 | 1.98 |
| Q06AA3 | RGN | regucalcin | 14 | 94.49 | 0.0034 | 1.71 |
| A0A5G2QAG3 | LAP3 | cytosol aminopeptidase isoform X1 | 14 | 79.04 | 0.0016 | 1.78 |
| P46410 | GLUL | glutamine synthetase isoform X1 | 13 | 70.53 | 0.0008 | 3.78 |
| A0A480R4L3 | KYNU | kynureninase isoform X1 | 13 | 50.36 | 0.0008 | 2.24 |
| A0A286ZZN9 | GLMS | glutamine-fructose-6-phosphate aminotransferase 1 | 13 | 58.72 | 0.0000 | 2.19 |
| P37111 | ACY1 | aminoacylase-1 | 12 | 86.53 | 0.0009 | 2.34 |
| Q6Q2C2 | EPHX2 | bifunctional epoxide hydrolase 2 | 12 | 54.89 | 0.0174 | 1.66 |
| A0A286ZRS0 | GSS | glutathione synthetase | 11 | 44.56 | 0.0071 | 1.91 |
| P28839 | LAP3 | xaa-Pro dipeptidase | 11 | 48.18 | 0.0023 | 2.64 |
| A0A4X1TBE8 | GSTM3 | glutathione S-transferase | 11 | 49.20 | 0.0023 | 1.72 |
| I3L804 | YARS1 | tyrosine-tRNA ligase, cytoplasmic isoform X1 | 11 | 41.54 | 0.0005 | 2.35 |
| I3LIM2 | UGDH | UDP-glucose 6-dehydrogenase | 11 | 45.24 | 0.0098 | 1.82 |
| Q02110 | HPD | 4-hydroxyphenylpyruvate dioxygenase | 11 | 55.49 | 0.0033 | 1.65 |
| D2SW95 | COPB1 | coatomer subunit beta | 11 | 40.14 | 0.0082 | 1.67 |
| A0A8D0YFM9 | GSR | glutathione reductase, mitochondrial | 10 | 58.65 | 0.0093 | 1.98 |
| A0A480NMC5 | DPP3 | dipeptidyl peptidase 3 isoform X1 | 10 | 53.31 | 0.0009 | 2.71 |
| O97788 | FABP4 | fatty acid-binding protein | 10 | 54.41 | 0.0011 | 1.67 |
| A0A4X1TZD5 | EEF1A | elongation factor 1-alpha 1 | 10 | 61.46 | 0.0008 | 2.28 |
| P03974 | VCP | transitional endoplasmic reticulum ATPase | 10 | 45.68 | 0.0014 | 2.04 |
| F1SNJ5 | AKR1D1 | 3-oxo-5-beta-steroid 4-dehydrogenase isoform X1 | 10 | 52.58 | 0.0026 | 1.75 |
| F1SIJ9 | PSAT1 | phosphoserine aminotransferase | 10 | 59.02 | 0.0008 | 1.63 |
| Enriched Pathway Description | p Value | p Adjust | q Value | GeneID | Protein Count |
|---|---|---|---|---|---|
| Carbon metabolism | 2.60 × 10−14 | 6.16 × 10−12 | 4.60 × 10−12 | CPS1, GLUD1, PC, PCCA, IDH2, PCCB, ACAT1, SUCLG2, MDH2, ACADS, CS, ACO1, DLD, FBP1, SHMT2, SUCLG1, GLDC, SDHA, MMUT, TALDO1, GLYCTK, AMT, DLST, HIBCH, HAO2, ALDOA, ALDOC, GCSH, RGN, PSAT1, PGK1, GPI, CAT, GAPDH, PHGDH, IDH1, ENO1, ESD, ALDOB, GCK, PGD | 41 |
| Ribosome | 7.46 × 10−8 | 8.84 × 10−6 | 6.60 × 10−7 | RPL26L1, RPS3, RPLP0, RPSA, RPL7, RPS4X, RPS3A, RPL9, RPL7A, RPL18, RPL30, RPS8, RPL4, RPS19, RPL13A, RPL19, RPL5, RPS7, RPS9, RPL10A, RPL27A, RPS18, RPS25, RPL24, RPS23, RPL8, RPS13, RPL37A, RPS21, RPL10L, RPL36, RPL35A, RPL23, RPS24, RPL6, RPS11, RPS17, RPS10, RPL22, RPS12 | 40 |
| Coronavirus disease | 3.13 × 10−2 | 1.49 × 10−1 | 1.11 × 10 | RPL26L1, RPS3, RPLP0, RPSA, RPL7, RPS4X, RPS3A, RPL9, STAT1, RPL7A, RPL18, RPL30, RPS8, RPL4, RPS19, RPL13A, RPL19, RPL5, RPS7, RPS9, RPL10A, RPL27A, RPS18, RPS25, FGB, RPL24, RPS23, RPL8, RPS13, RPL37A, RPS21, RPL10L, RPL36, RPL35A, C8A, RPL23, RPS24, RPL6, RPS11, RPS17, RPS10, RPL22, RPS12, FGG | 44 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martino, C.; Di Luca, A.; Bennato, F.; Ianni, A.; Passamonti, F.; Rampacci, E.; Henry, M.; Meleady, P.; Martino, G. Label-Free Quantitative Analysis of Pig Liver Proteome after Hepatitis E Virus Infection. Viruses 2024, 16, 408. https://doi.org/10.3390/v16030408
Martino C, Di Luca A, Bennato F, Ianni A, Passamonti F, Rampacci E, Henry M, Meleady P, Martino G. Label-Free Quantitative Analysis of Pig Liver Proteome after Hepatitis E Virus Infection. Viruses. 2024; 16(3):408. https://doi.org/10.3390/v16030408
Chicago/Turabian StyleMartino, Camillo, Alessio Di Luca, Francesca Bennato, Andrea Ianni, Fabrizio Passamonti, Elisa Rampacci, Michael Henry, Paula Meleady, and Giuseppe Martino. 2024. "Label-Free Quantitative Analysis of Pig Liver Proteome after Hepatitis E Virus Infection" Viruses 16, no. 3: 408. https://doi.org/10.3390/v16030408
APA StyleMartino, C., Di Luca, A., Bennato, F., Ianni, A., Passamonti, F., Rampacci, E., Henry, M., Meleady, P., & Martino, G. (2024). Label-Free Quantitative Analysis of Pig Liver Proteome after Hepatitis E Virus Infection. Viruses, 16(3), 408. https://doi.org/10.3390/v16030408