
Genomic Analyses of Major SARS-CoV-2 Variants Predicting Multiple Regions of Pathogenic and Transmissive Importance




Abstract
1. Introduction
2. Materials and Methods
2.1. Data Sources
2.2. Data Preparation
2.3. Statistical Analysis
2.3.1. Meta-CATS
2.3.2. HyPhy
2.3.3. MISTIC
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Phillips, N. The Coronavirus Is Here to Stay—Here’s What That Means. 2021. Available online: https://www.nature.com/articles/d41586-021-00396-2 (accessed on 18 October 2023).
- Kurland, C.G. Codon bias and gene expression. FEBS Lett. 1991, 285, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Quax, T.E.; Claassens, N.J.; Söll, D.; van der Oost, J. Codon Bias as a Means to Fine-Tune Gene Expression. Mol. Cell 2015, 59, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Hershberg, R.; Petrov, D.A. Selection on codon bias. Annu. Rev. Genet. 2008, 42, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Stoletzki, N.; Eyre-Walker, A. Synonymous codon usage in Escherichia coli: Selection for translational accuracy. Mol. Biol. Evol. 2007, 24, 374–381. [Google Scholar] [CrossRef]
- Qian, W.; Qian, W.; Yang, J.-R.; Pearson, N.M.; Maclean, C.; Zhang, J. Balanced codon usage optimizes eukaryotic translational efficiency. PLoS Genet. 2012, 8, e1002603. [Google Scholar] [CrossRef]
- Dana, A.; Tuller, T. The effect of tRNA levels on decoding times of mRNA codons. Nucleic Acids Res. 2014, 42, 9171–9181. [Google Scholar] [CrossRef]
- Pechmann, S.; Chartron, J.W.; Frydman, J. Local slowdown of translation by nonoptimal codons promotes nascent-chain recognition by SRP in vivo. Nat. Struct. Mol. Biol. 2014, 21, 1100–1105. [Google Scholar] [CrossRef] [PubMed]
- Hia, F.; Yang, S.F.; Shichino, Y.; Yoshinaga, M.; Murakawa, Y.; Vandenbon, A.; Fukao, A.; Fujiwara, T.; Landthaler, M.; Natsume, T.; et al. Codon bias confers stability to human mRNAs. EMBO Rep. 2019, 20, e48220. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.V.; Payne, J.L. Mutation bias interacts with composition bias to influence adaptive evolution. PLoS Comput. Biol. 2020, 16, e1008296. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.B.; Miller, J.B.; Hippen, A.A.; Wright, S.M.; Morris, C.; Ridge, P.G. Human viruses have codon usage biases that match highly expressed proteins in the tissues they infect. Biomed. Genet. Genom. 2017, 2, 1–5. [Google Scholar] [CrossRef]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Alexaki, A.; Kames, J.; Holcomb, D.D.; Athey, J.; Santana-Quintero, L.V.; Lam, P.V.N.; Hamasaki-Katagiri, N.; Osipova, E.; Simonyan, V.; Bar, H.; et al. Codon and Codon-Pair Usage Tables (CoCoPUTs): Facilitating Genetic Variation Analyses and Recombinant Gene Design. J. Mol. Biol. 2019, 431, 2434–2441. [Google Scholar] [CrossRef] [PubMed]
- Pickett, B.E.; Liu, M.; Sadat, E.; Squires, R.; Noronha, J.; He, S.; Jen, W.; Zaremba, S.; Gu, Z.; Zhou, L.; et al. Metadata-driven comparative analysis tool for sequences (meta-CATS): An automated process for identifying significant sequence variations that correlate with virus attributes. Virology 2013, 447, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Pond, S.L.K. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef] [PubMed]
- Simonetti, F.L.; Teppa, E.; Chernomoretz, A.; Nielsen, M.; Buslje, C.M. MISTIC: Mutual information server to infer coevolution. Nucleic Acids Res. 2013, 41, W8–W14. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Alonso, A.M.; Diambra, L. SARS-CoV-2 Codon Usage Bias Downregulates Host Expressed Genes With Similar Codon Usage. Front. Cell Dev. Biol. 2020, 8, 831. [Google Scholar] [CrossRef]
- Johnson, B.A.; Zhou, Y.; Lokugamage, K.G.; Vu, M.N.; Bopp, N.; Crocquet-Valdes, P.A.; Kalveram, B.; Schindewolf, C.; Liu, Y.; Scharton, D.; et al. Nucleocapsid mutations in SARS-CoV-2 augment replication and pathogenesis. PLoS Pathog. 2022, 18, e1010627. [Google Scholar] [CrossRef]
- Yang, M.; Derbyshire, M.K.; Yamashita, R.A.; Marchler-Bauer, A. NCBI’s Conserved Domain Database and Tools for Protein Domain Analysis. Curr. Protoc. Bioinformatics 2020, 69, e90. [Google Scholar] [CrossRef]
- Ye, Q.; West, A.M.V.; Silletti, S.; Corbett, K.D. Architecture and self-assembly of the SARS-CoV-2 nucleocapsid protein. Protein Sci. 2020, 29, 1890–1901. [Google Scholar] [CrossRef]
- Kohyama, M.; Suzuki, T.; Nakai, W.; Ono, C.; Matsuoka, S.; Iwatani, K.; Liu, Y.; Sakai, Y.; Nakagawa, A.; Tomii, K.; et al. SARS-CoV-2 ORF8 is a viral cytokine regulating immune responses. Int. Immunol. 2023, 35, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Vinjamuri, S.; Li, L.; Bouvier, M. SARS-CoV-2 ORF8: One protein, seemingly one structure, and many functions. Front. Immunol. 2022, 13, 6459. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, A.M.; Singh, I.; Joshi, M.; Patel, A.; Joshi, C. Defective ORF8 dimerization in SARS-CoV-2 delta variant leads to a better adaptive immune response due to abrogation of ORF8-MHC1 interaction. Mol. Divers. 2023, 27, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Tarres-Freixas, F.; Trinité, B.; Pons-Grífols, A.; Romero-Durana, M.; Riveira-Muñoz, E.; Ávila-Nieto, C.; Pérez, M.; Garcia-Vidal, E.; Perez-Zsolt, D.; Muñoz-Basagoiti, J.; et al. Heterogeneous Infectivity and Pathogenesis of SARS-CoV-2 Variants Beta, Delta and Omicron in Transgenic K18-hACE2 and Wildtype Mice. Front. Microbiol. 2022, 13, 840757. [Google Scholar] [CrossRef]
- Shuai, H.; Chan, J.F.-W.; Hu, B.; Chai, Y.; Yuen, T.T.-T.; Yin, F.; Huang, X.; Yoon, C.; Hu, J.-C.; Liu, H.; et al. Attenuated replication and pathogenicity of SARS-CoV-2 B.1.1.529 Omicron. Nature 2022, 603, 693–699. [Google Scholar] [CrossRef]
- Dao, T.L.; Hoang, V.T.; Colson, P.; Lagier, J.C.; Million, M.; Raoult, D.; Levasseur, A.; Gautret, P. SARS-CoV-2 Infectivity and Severity of COVID-19 According to SARS-CoV-2 Variants: Current Evidence. J. Clin. Med. 2021, 10, 2635. [Google Scholar] [CrossRef]
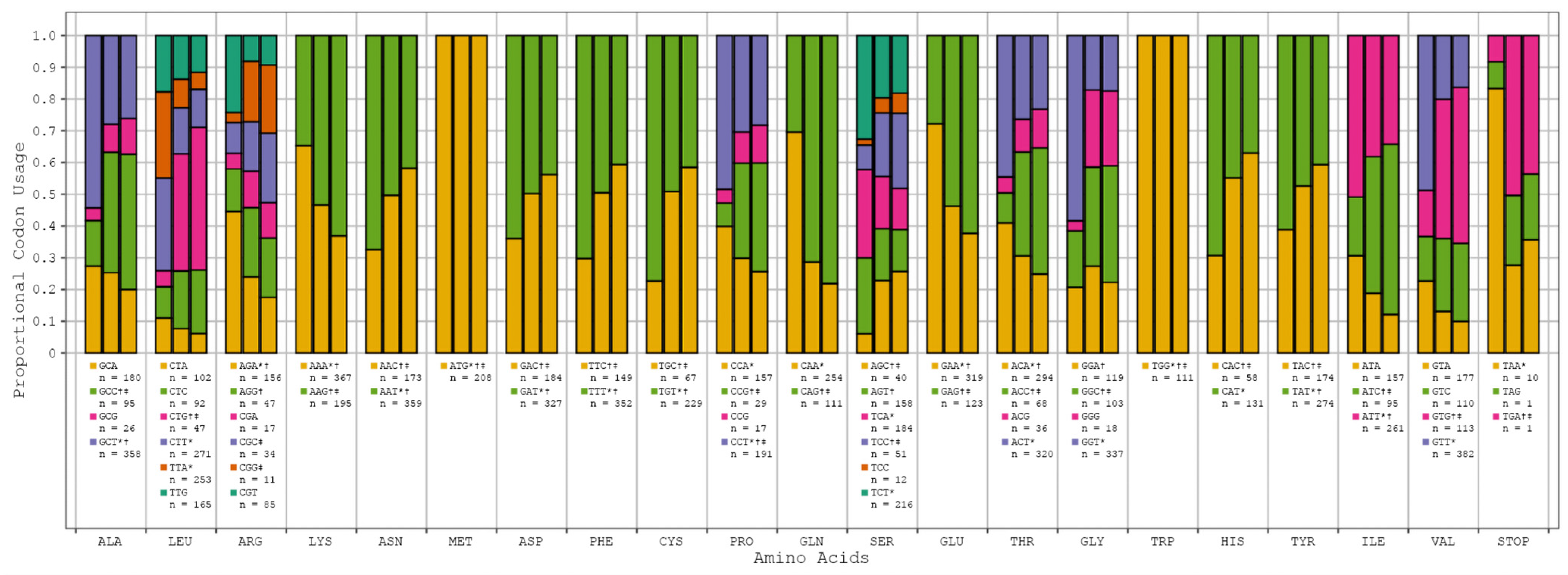
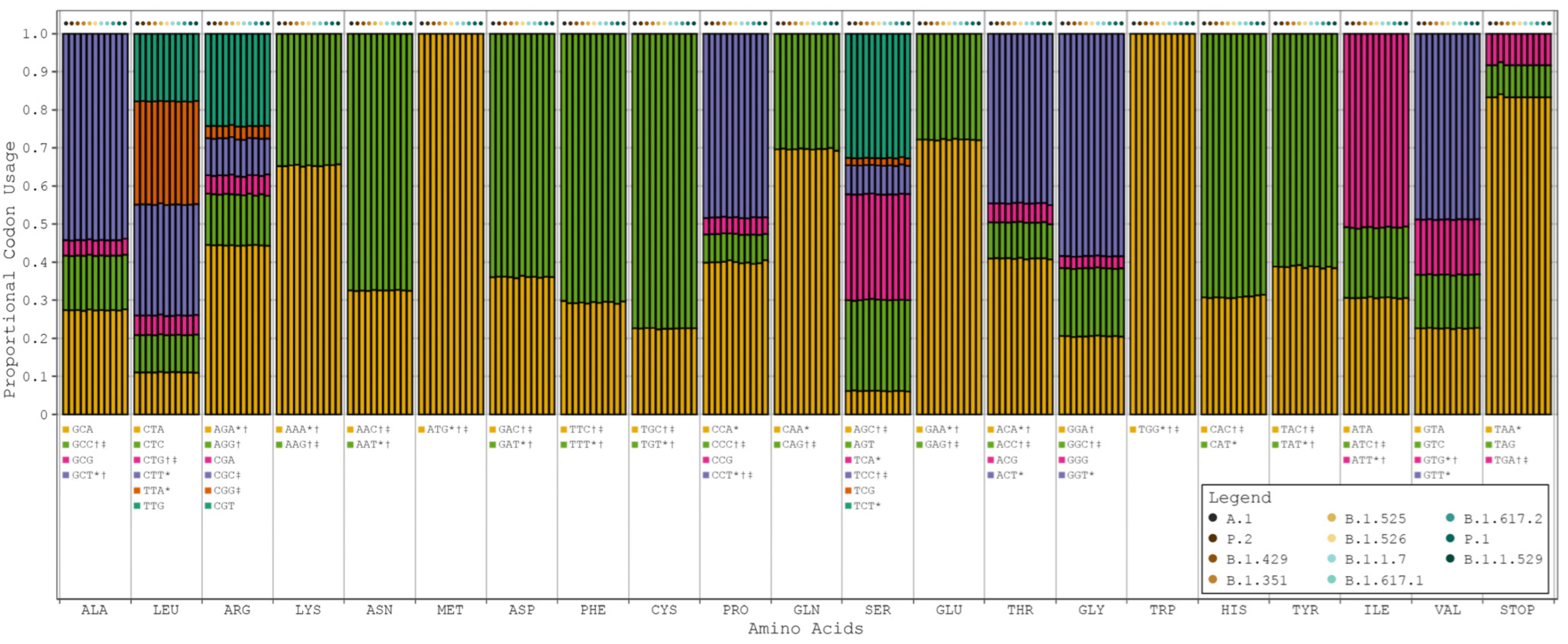


{kind=link}
{kind=link}
{kind=link}
| Change Type | N (%) | Number Synonymous (%) | Number Nonsynonymous (%) | Number Deletion (%) |
|---|---|---|---|---|
| Toward SARS-CoV-2 | 49/241 (20%) | 34/49 (67%) | 17/49 (33%) | 0/49 (0%) |
| Toward HG | 30/241 (12%) | 4/30 (13%) | 26/30 (87%) | 0/30 (0%) |
| Toward HPT | 10/241 (4%) | 7/10 (70%) | 3/10 (30%) | 0/10 (0%) |
| Toward non bias | 70/241 (29%) | 8/70 (11%) | 23/70 (33%) | 39/70 (56%) |
| Mutation by Codon Position | N (%) |
|---|---|
| 1 | 50/241 (21%) |
| 2 | 71/241 (30%) |
| 3 | 61/241 (25%) |
| 1/2 | 6/241 (2%) |
| 2/3 | 8/241 (3%) |
| 1/2/3 | 45/241 (19%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brugger, S.W.; Grose, J.H.; Decker, C.H.; Pickett, B.E.; Davis, M.F. Genomic Analyses of Major SARS-CoV-2 Variants Predicting Multiple Regions of Pathogenic and Transmissive Importance. Viruses 2024, 16, 276. https://doi.org/10.3390/v16020276
Brugger SW, Grose JH, Decker CH, Pickett BE, Davis MF. Genomic Analyses of Major SARS-CoV-2 Variants Predicting Multiple Regions of Pathogenic and Transmissive Importance. Viruses. 2024; 16(2):276. https://doi.org/10.3390/v16020276
Chicago/Turabian StyleBrugger, Steven W., Julianne H. Grose, Craig H. Decker, Brett E. Pickett, and Mary F. Davis. 2024. "Genomic Analyses of Major SARS-CoV-2 Variants Predicting Multiple Regions of Pathogenic and Transmissive Importance" Viruses 16, no. 2: 276. https://doi.org/10.3390/v16020276
APA StyleBrugger, S. W., Grose, J. H., Decker, C. H., Pickett, B. E., & Davis, M. F. (2024). Genomic Analyses of Major SARS-CoV-2 Variants Predicting Multiple Regions of Pathogenic and Transmissive Importance. Viruses, 16(2), 276. https://doi.org/10.3390/v16020276