
Enzootic Circulation, Massive Gull Mortality and Poultry Outbreaks during the 2022/2023 High-Pathogenicity Avian Influenza H5N1 Season in the Czech Republic
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
3. Results
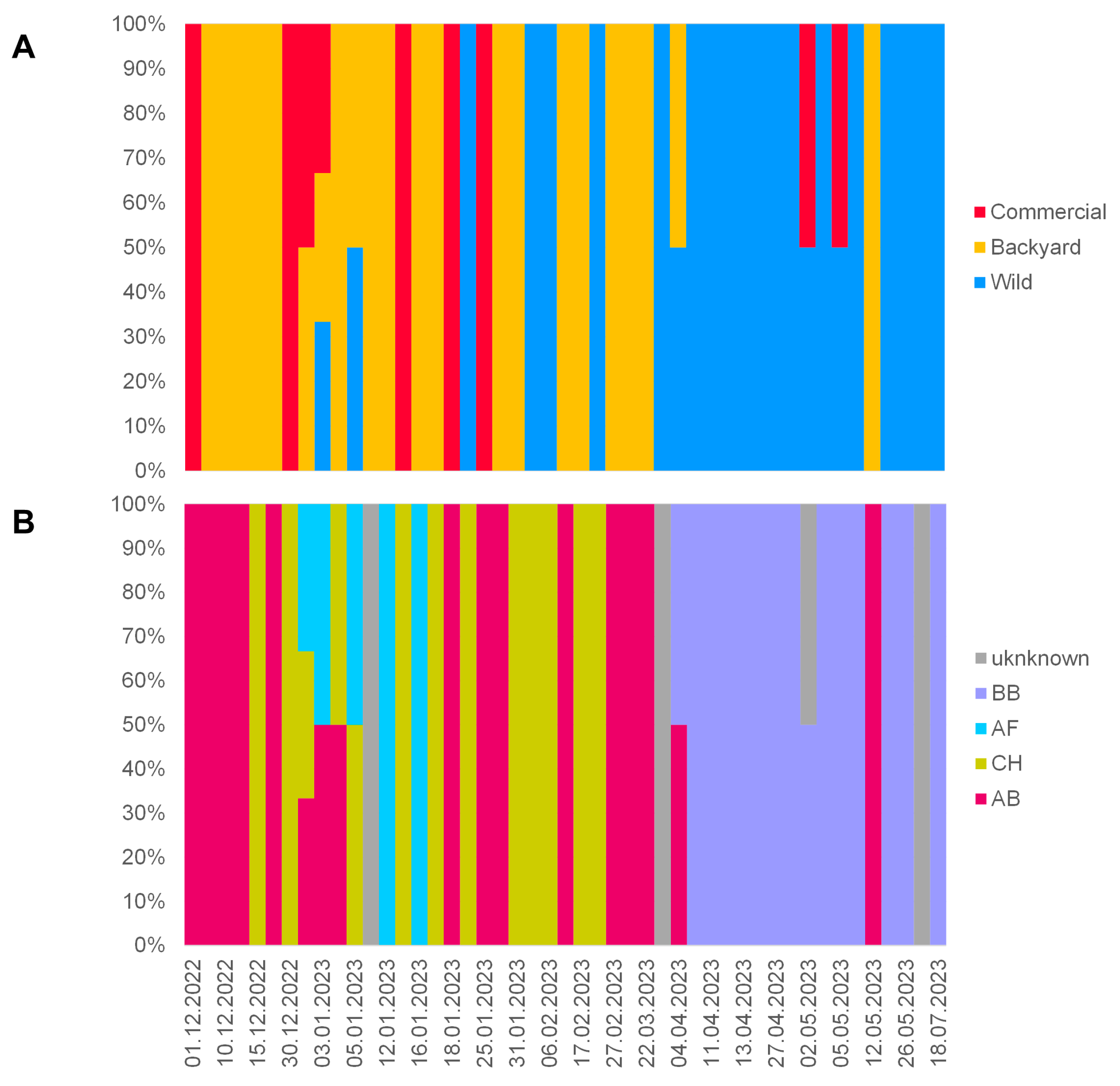
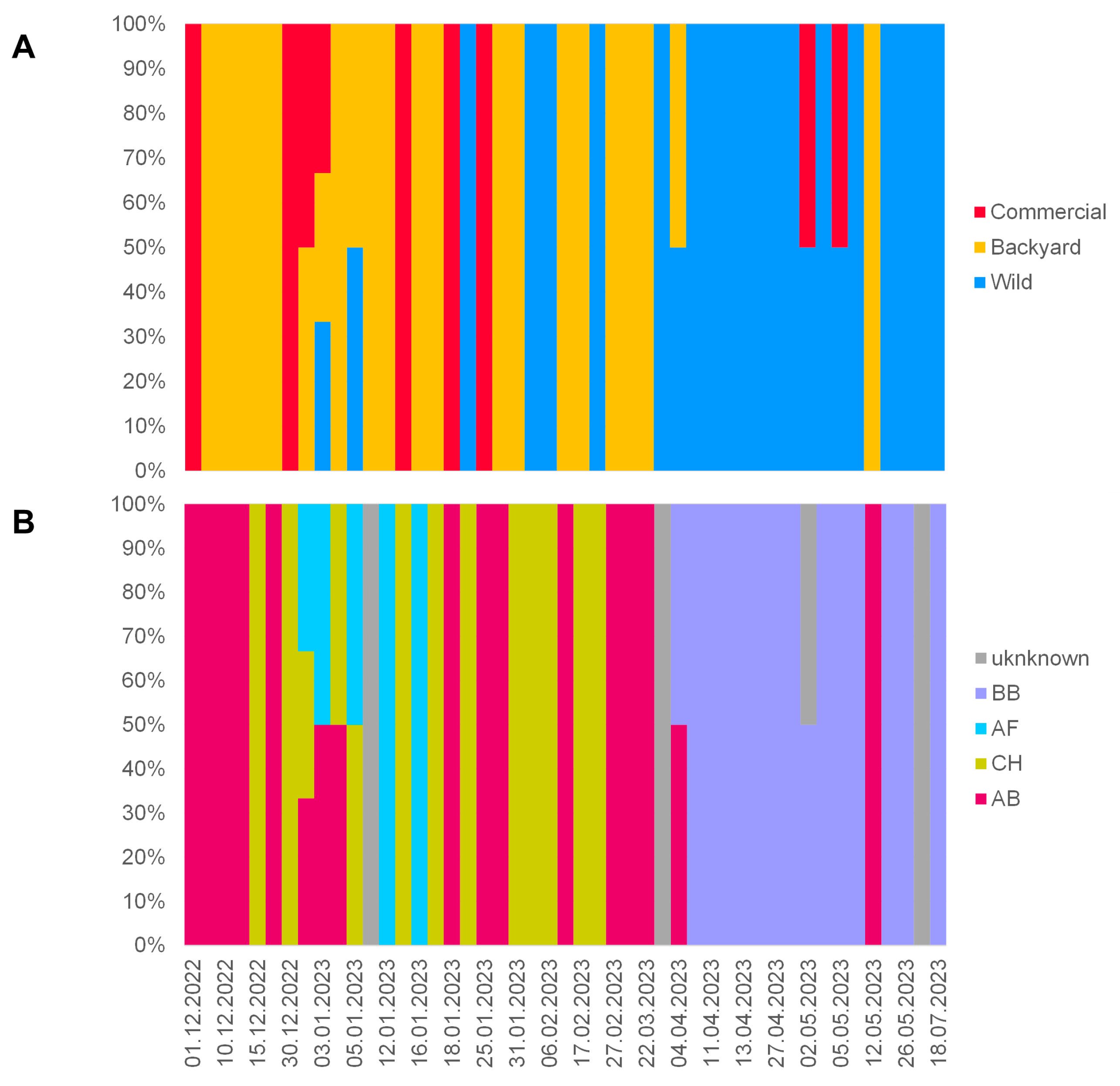
3.1. Overview of the 2022/2023 H5N1 HPAI Season in the Czech Republic
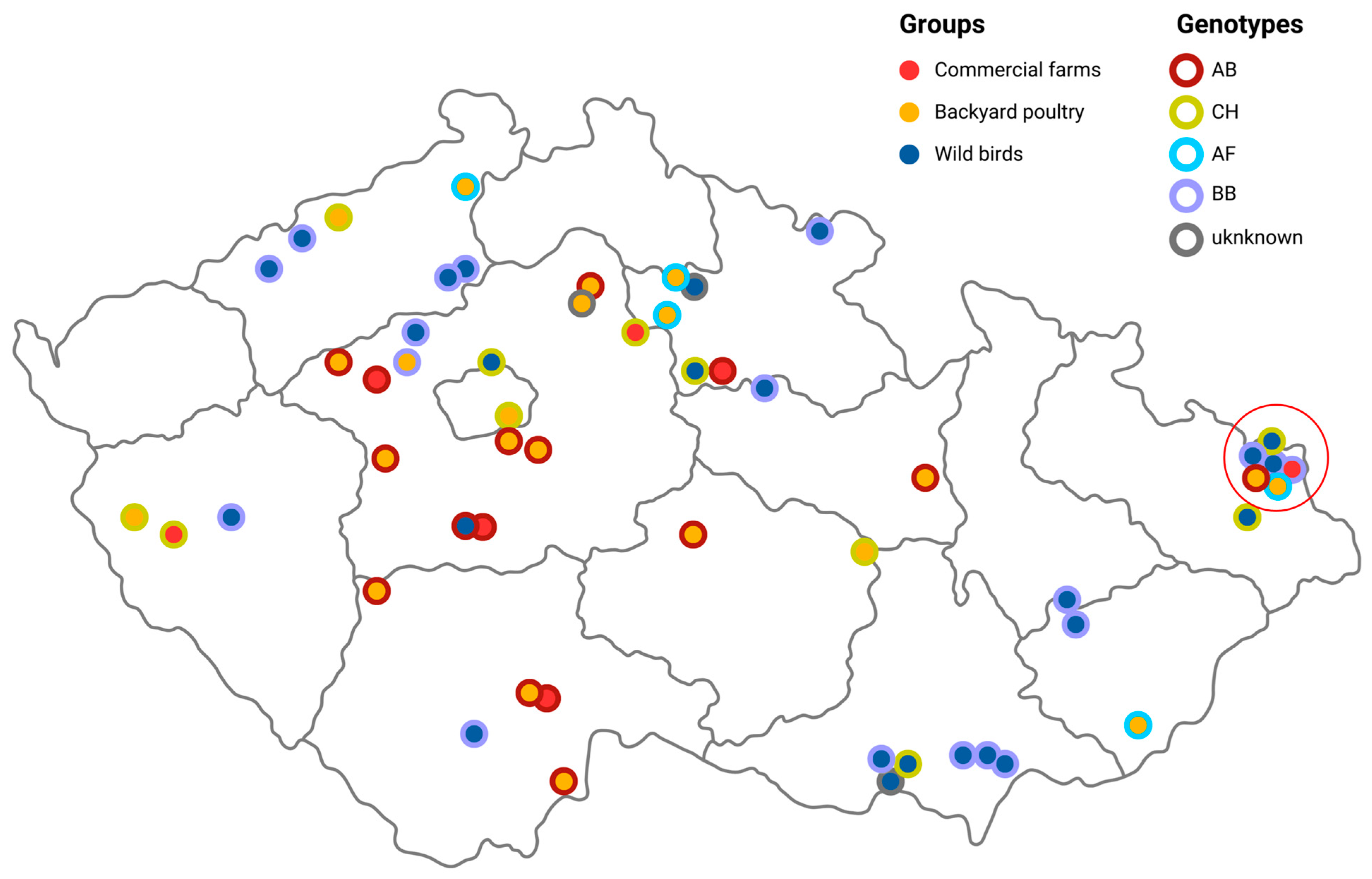
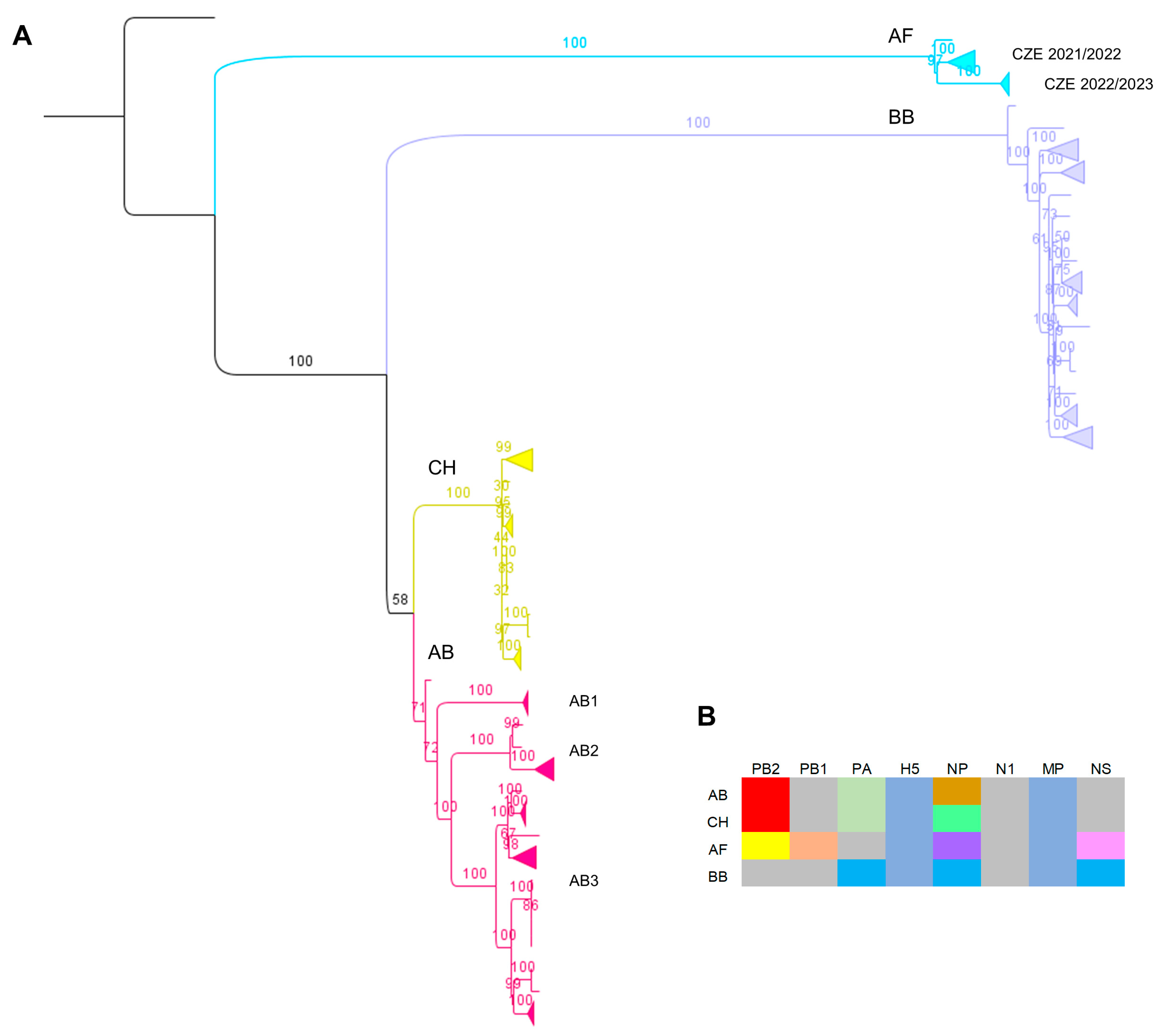
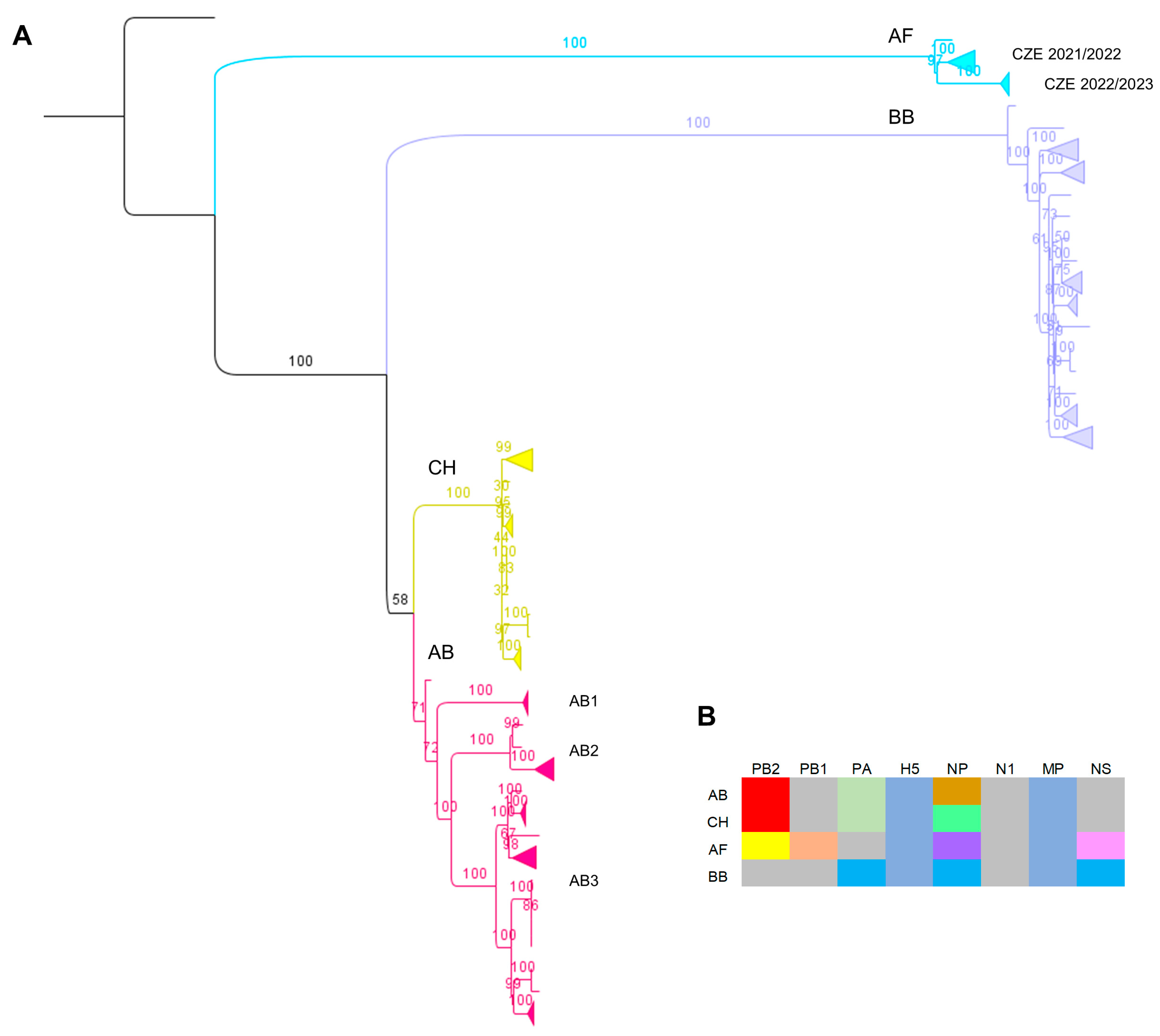
3.2. Genotyping and Molecular Characterization of the Detected H5N1 Viruses
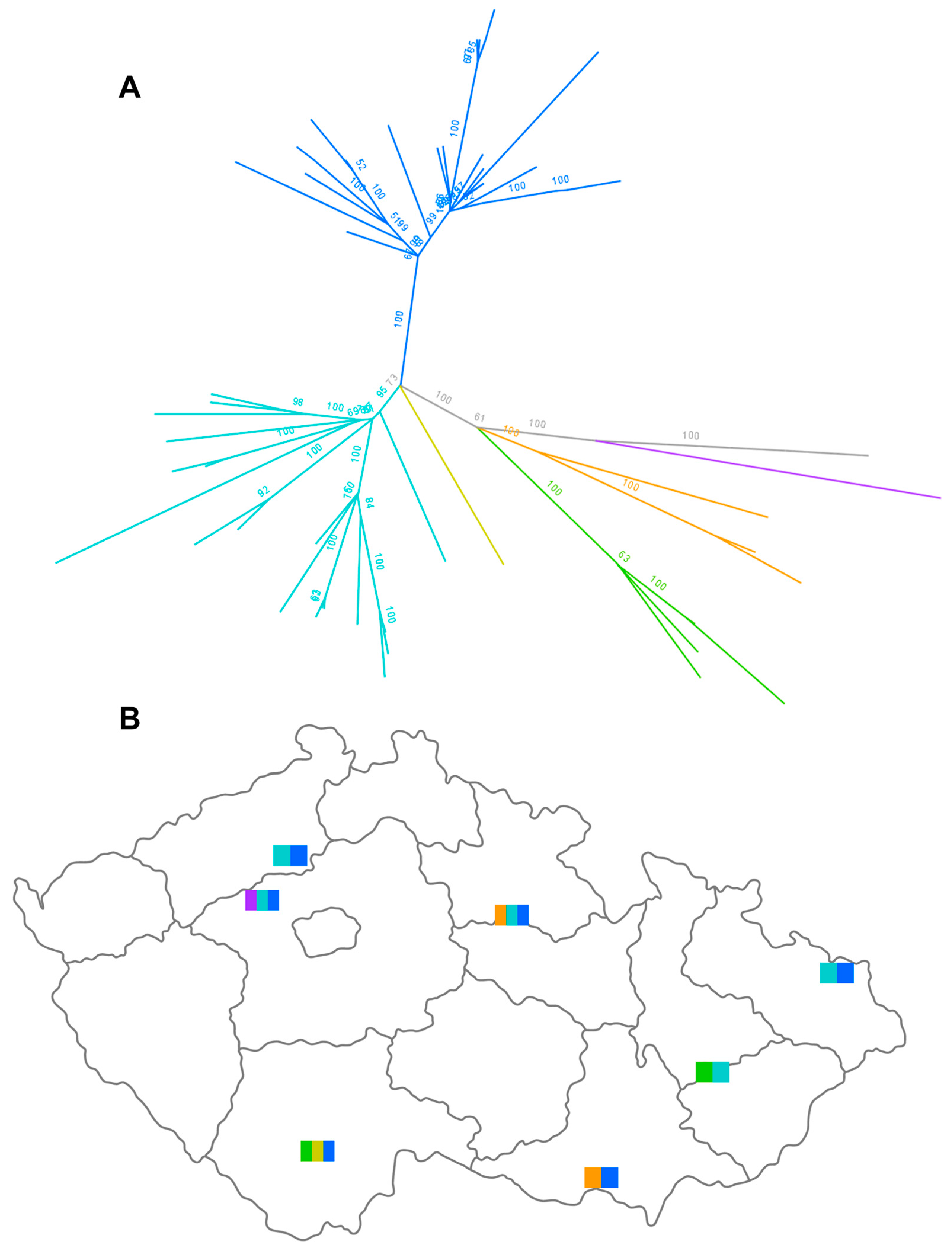
3.3. Molecular Epizootology of the Czech 2022/2023 H5N1 Viruses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- EFSA (European Food Safety Authority); ECDC (European Centre for Disease Prevention and Control); EURL (European Reference Laboratory for Avian Influenza); Adlhoch, C.; Fusaro, A.; Gonzales, J.L.; Kuiken, T.; Marangon, S.; Niqueux, É.; Staubach, C.; et al. Scientific report: Avian influenza overview June–September 2022. EFSA J. 2022, 20, 7597. [Google Scholar] [CrossRef]
- Xie, R.; Edwards, K.M.; Wille, M.; Wei, X.; Wong, S.S.; Zanin, M.; El-Shesheny, R.; Ducatez, M.; Poon, L.L.M.; Kayali, G.; et al. The episodic resurgence of highly pathogenic avian influenza H5 virus. Nature 2023, 622, 810–817. [Google Scholar] [CrossRef] [PubMed]
- Munster, V.J.; Baas, C.; Lexmond, P.; Waldenström, J.; Wallensten, A.; Fransson, T.; Rimmelzwaan, G.F.; Beyer, W.E.; Schutten, M.; Olsen, B.; et al. Spatial, temporal, and species variation in prevalence of influenza A viruses in wild migratory birds. PLoS Pathog. 2007, 3, 5. [Google Scholar] [CrossRef]
- Cattoli, G.; Fusaro, A.; Monne, I.; Capua, I. H5N1 Virus Evolution in Europe-An Updated Overview. Viruses 2009, 1, 1351–1363. [Google Scholar] [CrossRef] [PubMed]
- Lewis, N.S.; Banyard, A.C.; Whittard, E.; Karibayev, T.; Al Kafagi, T.; Chvala, I.; Byrne, A.; Meruyert Akberovna, S.; King, J.; Harder, T.; et al. Emergence and spread of novel H5N8, H5N5 and H5N1 clade 2.3.4.4 highly pathogenic avian influenza in 2020. Emerg. Microbes Infect. 2021, 10, 148–151. [Google Scholar] [CrossRef] [PubMed]
- Pohlmann, A.; King, J.; Fusaro, A.; Zecchin, B.; Banyard, A.C.; Brown, I.H.; Byrne, A.M.P.; Beerens, N.; Liang, Y.; Heutink, R.; et al. Has Epizootic Become Enzootic? Evidence for a Fundamental Change in the Infection Dynamics of Highly Pathogenic Avian Influenza in Europe, 2021. mBio 2022, 13, e0060922. [Google Scholar] [CrossRef] [PubMed]
- EFSA (European Food Safety Authority); ECDC (European Centre for Disease Prevention and Control); EURL (European Reference Laboratory for Avian Influenza); Brown, I.; Mulatti, P.; Smietanka, K.; Staubach, C.; Willeberg, P.; Adlhoch, C.; Candiani, D.; et al. Scientific report on the avian influenza overview October 2016–August 2017. EFSA J. 2017, 15, 5018. [Google Scholar] [CrossRef]
- EFSA (European Food Safety Authority); ECDC (European Centre for Disease Prevention and Control); EURL (European Reference Laboratory for Avian Influenza); Adlhoch, C.; Fusaro, A.; Gonzales, J.L.; Kuiken, T.; Marangon, S.; Niqueux, É.; Staubach, C.; et al. Scientific report: Avian influenza overview March–June 2022. EFSA J. 2022, 20, 7415. [Google Scholar] [CrossRef]
- EFSA (European Food Safety Authority); ECDC (European Centre for Disease Prevention and Control); EURL (European Reference Laboratory for Avian Influenza); Adlhoch, C.; Fusaro, A.; Gonzales, J.L.; Kuiken, T.; Marangon, S.; Niqueux, É.; Staubach, C.; et al. Scientific report: Avian influenza overview August–December 2020. EFSA J. 2020, 18, 6379. [Google Scholar] [CrossRef]
- EFSA (European Food Safety Authority); ECDC (European Centre for Disease Prevention and Control); EURL (European Reference Laboratory for Avian Influenza); Adlhoch, C.; Fusaro, A.; Gonzales, J.L.; Kuiken, T.; Marangon, S.; Niqueux, É.; Staubach, C.; et al. Scientific report: Avian influenza overview May–September 2021. EFSA J. 2022, 20, 7122. [Google Scholar] [CrossRef]
- Banyard, A.C.; Lean, F.Z.X.; Robinson, C.; Howie, F.; Tyler, G.; Nisbet, C.; Seekings, J.; Meyer, S.; Whittard, E.; Ashpitel, H.F.; et al. Detection of Highly Pathogenic Avian Influenza Virus H5N1 Clade 2.3.4.4b in Great Skuas: A Species of Conservation Concern in Great Britain. Viruses 2022, 14, 212. [Google Scholar] [CrossRef]
- Rijks, J.M.; Leopold, M.F.; Kühn, S.; In ‘t Veld, R.; Schenk, F.; Brenninkmeijer, A.; Lilipaly, S.J.; Ballmann, M.Z.; Kelder, L.; de Jong, J.W.; et al. Mass Mortality Caused by Highly Pathogenic Influenza A(H5N1) Virus in Sandwich Terns, the Netherlands, 2022. Emerg. Infect. Dis. 2022, 28, 2538–2542. [Google Scholar] [CrossRef] [PubMed]
- Pohlmann, A.; Stejskal, O.; King, J.; Bouwhuis, S.; Packmor, F.; Ballstaedt, E.; Hälterlein, B.; Hennig, V.; Stacker, L.; Graaf, A.; et al. Mass mortality among colony-breeding seabirds in the German Wadden Sea in 2022 due to distinct genotypes of HPAIV H5N1 clade 2.3.4.4b. J. Gen. Virol. 2023, 104, 001834. [Google Scholar] [CrossRef] [PubMed]
- Lane, J.V.; Jeglinski, J.W.E.; Avery-Gomm, S.; Ballstaedt, E.; Banyard, A.C.; Barychka, T.; Brown, I.H.; Brugger, B.; Burt, T.V.; Careen, N.; et al. High pathogenicity avian influenza (H5N1) in Northern Gannets (Morus bassanus): Global spread, clinical signs and demographic consequences. Ibis 2023. [CrossRef]
- Camphuysen, K.; Gear, S. Great Skuas and Northern Gannets on Foula, summer 2022—An unprecedented, H5N1 related massacre. NIOZ 2022. [CrossRef]
- Knief, U.; Bregnballe, T.; Alfarwi, I.; Ballmann, M.; Brenninkmeijer, A.; Bzoma, S.; Chabrolle, A.; Dimmlich, J.; Engel, E.; Fijn, R.; et al. Highly pathogenic avian influenza causes mass mortality in Sandwich tern (Thalasseus sandvicensis) breeding colonies across northwestern Europe. bioRxiv 2023. [Google Scholar] [CrossRef]
- EFSA (European Food Safety Authority); ECDC (European Centre for Disease Prevention and Control); EURL (European Reference Laboratory for Avian Influenza); Adlhoch, C.; Fusaro, A.; Gonzales, J.L.; Kuiken, T.; Marangon, S.; Stahl, K.; Niqueux, É.; et al. Scientific report: Avian influenza overview December 2022–March 2023. EFSA J. 2023, 21, 7917. [Google Scholar] [CrossRef]
- Nagy, A.; Černíková, L.; Kunteová, K.; Dirbáková, Z.; Thomas, S.S.; Slomka, M.J.; Dán, Á.; Varga, T.; Máté, M.; Jiřincová, H.; et al. A universal RT-qPCR assay for "One Health" detection of influenza A viruses. PLoS ONE 2021, 16, e0244669. [Google Scholar] [CrossRef]
- Slomka, M.J.; Pavlidis, T.; Banks, J.; Shell, W.; McNally, A.; Essen, S.; Brown, I.H. Validated H5 Eurasian real-time reverse transcriptase-polymerase chain reaction and its application in H5N1 outbreaks in 2005–2006. Avian Dis. 2007, 51, 373–377. [Google Scholar] [CrossRef]
- Payungporn, S.; Chutinimitkul, S.; Chaisingh, A.; Damrongwantanapokin, S.; Buranathai, C.; Amonsin, A.; Theamboonlers, A.; Poovorawan, Y. Single step multiplex real-time RT-PCR for H5N1 influenza A virus detection. J. Virol. Methods 2006, 131, 143–147. [Google Scholar] [CrossRef]
- James, J.; Seekings, A.H.; Skinner, P.; Purchase, K.; Mahmood, S.; Brown, I.H.; Hansen, R.D.E.; Banyard, A.C.; Reid, S.M. Rapid and sensitive detection of high pathogenicity Eurasian clade 2.3.4.4b avian influenza viruses in wild birds and poultry. J. Virol. Methods 2022, 301, 114454. [Google Scholar] [CrossRef]
- Arctic Network. Available online: https://web.archive.org/web/20221021094307/https://artic.network/ (accessed on 17 February 2022).
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Sahlin, K.; Lim, M.C.W.; Prost, S. NGSpeciesID: DNA barcode and amplicon consensus generation from long-read sequencing data. Ecol. Evol. 2021, 11, 1392–1398. [Google Scholar] [CrossRef] [PubMed]
- Vierstraete, A.R.; Braeckman, B.P. Amplicon_sorter: A tool for reference-free amplicon sorting based on sequence similarity and for building consensus sequences. Ecol. Evol. 2022, 12, e8603. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Kazunori, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Hatcher, E.L.; Zhdanov, S.A.; Bao, Y.; Blinkova, O.; Nawrocki, E.P.; Ostapchuck, Y.; Schäffer, A.A.; Brister, J.R. Virus Variation Resource—Improved response to emergent viral outbreaks. Nucleic Acids Res. 2017, 45, D482–D490. [Google Scholar] [CrossRef]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef] [PubMed]
- Trifinopoulos, J.; Nguyen, L.T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- EFSA (European Food Safety Authority); ECDC (European Centre for Disease Prevention and Control); EURL (European Reference Laboratory for Avian Influenza); Adlhoch, C.; Fusaro, A.; Gonzales, J.L.; Kuiken, T.; Mirinaviciute, G.; Niqueux, É.; Stahl, K.; et al. Scientific report: Avian influenza overview March–April 2023. EFSA J. 2023, 21, 8039. [Google Scholar] [CrossRef]
- Suttie, A.; Deng, Y.M.; Greenhill, A.R.; Dussart, P.; Horwood, P.F.; Karlsson, E.A. Inventory of molecular markers affecting biological characteristics of avian influenza A viruses. Virus Genes 2019, 6, 739–768. [Google Scholar] [CrossRef]
- Pinto, R.M.; Bakshi, S.; Lytras, S.; Zakaria, M.K.; Swingler, S.; Worrell, J.C.; Herder, V.; Hargrave, K.E.; Varjak, M.; Cameron-Ruiz, N.; et al. BTN3A3 evasion promotes the zoonotic potential of influenza A viruses. Nature 2023, 619, 338–347. [Google Scholar] [CrossRef] [PubMed]
- EFSA (European Food Safety Authority); ECDC (European Centre for Disease Prevention and Control); EURL (European Reference Laboratory for Avian Influenza); Adlhoch, C.; Fusaro, A.; Gonzales, J.L.; Kuiken, T.; Melidou, A.; Mirinavičiūtė, G.; Niqueux, É.; et al. Scientific report: Avian influenza overview April–June 2023. EFSA J. 2023, 21, 8191. [Google Scholar] [CrossRef]
- Fusaro, A.; Zecchin, B.; Giussani, E.; Palumbo, E.; Agüero-García, M.; Bachofen, C.; Bálint, Á.; Banihashem, F.; Banyard, A.C.; Beerens, N.; et al. High pathogenic avian influenza A(H5) viruses of clade 2.3.4.4b in Europe—why trends of virus evolution are more difficult to predict. under review.
- Nagy, A.; Stará, M.; Černíková, L.; Hofmannová, L.; Sedlák, K. Genotype Diversity, Wild Bird-to-Poultry Transmissions, and Farm-to-Farm Carryover during the Spread of the Highly Pathogenic Avian Influenza H5N1 in the Czech Republic in 2021/2022. Viruses 2023, 15, 293. [Google Scholar] [CrossRef]
- Nagy, A.; Černíková, L.; Stará, M.; Hofmannová, L.; Sedlák, K. Genotype Uniformity, Wild Bird-to-Poultry Transmissions, and Farm-to-Farm Carryover during the Spread of the Highly Pathogenic Avian Influenza H5N8 in the Czech Republic in 2021. Viruses 2022, 14, 1411. [Google Scholar] [CrossRef] [PubMed]
- Bouwstra, R.J.; Koch, G.; Heutink, R.; Harders, F.; van der Spek, A.; Elbers, A.R.; Bossers, A. Phylogenetic analysis of highly pathogenic avian influenza A(H5N8) virus outbreak strains provides evidence for four separate introductions and one between-poultry farm transmission in the Netherlands, November 2014. Eurosurveillance 2015, 20, 21174. [Google Scholar] [CrossRef]
- Śmietanka, K.; Świętoń, E.; Kozak, E.; Wyrostek, K.; Tarasiuk, K.; Tomczyk, G.; Konopka, B.; Welz, M.; Domańska-Blicharz, K.; Niemczuk, K. Highly Pathogenic Avian Influenza H5N8 in Poland in 2019–2020. J. Vet. Res. 2020, 64, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, D.; Brouwer, A.; Goujgoulova, G.; Ellis, R.; Seekings, J.; Brown, I.H.; Lewis, N.S. Regional Transmission and Reassortment of 2.3.4.4b Highly Pathogenic Avian Influenza (HPAI) Viruses in Bulgarian Poultry 2017/18. Viruses 2020, 12, 605. [Google Scholar] [CrossRef]
- Lambert, S.; Durand, B.; Andraud, M.; Delacourt, R.; Scoizec, A.; Le Bouquin, S.; Rautureau, S.; Bauzile, B.; Guinat, C.; Fourtune, L.; et al. Two major epidemics of highly pathogenic avian influenza virus H5N8 and H5N1 in domestic poultry in France, 2020-2022. Transbound. Emerg. Dis. 2022, 69, 3160–3166. [Google Scholar] [CrossRef]
- Filaire, F.; Lebre, L.; Foret-Lucas, C.; Vergne, T.; Daniel, P.; Lelièvre, A.; De Barros, A.; Jbenyeni, A.; Bolon, P.; Paul, M.; et al. Highly Pathogenic Avian Influenza A(H5N8) Clade 2.3.4.4b Virus in Dust Samples from Poutry Farms, France, 2021. Emerg. Infect. Dis. 2022, 28, 1446–1450. [Google Scholar] [CrossRef]
- Velkers, F.C.; Manders, T.T.M.; Vernooij, J.C.M.; Stahl, J.; Slaterus, R.; Stegeman, J.A. Association of wild bird densities around poultry farms with the risk of highly pathogenic avian influenza virus subtype H5N8 outbreaks in the Netherlands, 2016. Transbound Emerg Dis 2021, 68, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Soll, L.; Dugan, V.; Runstadler, J.; Happ, G.; Slemons, R.D.; Taubenberger, J.K. Examining the hemagglutinin subtype diversity among wild duck-origin influenza A viruses using ethanol-fixed cloacal swabs and a novel RT-PCR method. Virology 2008, 375, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Hill, N.J.; Takekawa, J.Y.; Cardona, C.J.; Meixell, B.W.; Ackerman, J.T.; Runstadler, J.A.; Boyce, W.M. Cross-seasonal patterns of avian influenza virus in breeding and wintering migratory birds: A flyway perspective. Vector Borne Zoonotic Dis. 2012, 12, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, L.L.; Kelly, T.R.; Plancarte, M.; Schobel, S.; Lin, X.; Dugan, V.G.; Wentworth, D.E.; Boyce, W.M. Avian influenza: Mixed infections and missing viruses. Viruses 2013, 5, 1964–1977. [Google Scholar] [CrossRef] [PubMed]
- Nagy, A.; Černíková, L.; Jiřincová, H.; Havlíčková, M.; Horníčková, J. Local-scale diversity and between-year “frozen evolution” of avian influenza A viruses in nature. PLoS ONE 2014, 30, e103053. [Google Scholar] [CrossRef] [PubMed]
- Hill, N.J.; Bishop, M.A.; Trovão, N.S.; Ineson, K.M.; Schaefer, A.L.; Puryear, W.B.; Zhou, K.; Foss, A.D.; Clark, D.E.; MacKenzie, K.G.; et al. Ecological divergence of wild birds drives avian influenza spillover and global spread. PLoS Pathog. 2022, 18, e1010062. [Google Scholar] [CrossRef] [PubMed]
- Cepak, J.; Klvana, P.; Skopek, J.; Schropfer, L.; Jelinek, M.; Horak, D.; Formanek, J.; Zarybnicky, J. Czech and Slovak Bird Migration Atlas, 1st ed.; Aventinum: Prague, Czech Republic, 2008; pp. 217–221. [Google Scholar]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagy, A.; Stará, M.; Černíková, L.; Kličková, E.; Horák, O.; Hofmannová, L.; Sedlák, K. Enzootic Circulation, Massive Gull Mortality and Poultry Outbreaks during the 2022/2023 High-Pathogenicity Avian Influenza H5N1 Season in the Czech Republic. Viruses 2024, 16, 221. https://doi.org/10.3390/v16020221
Nagy A, Stará M, Černíková L, Kličková E, Horák O, Hofmannová L, Sedlák K. Enzootic Circulation, Massive Gull Mortality and Poultry Outbreaks during the 2022/2023 High-Pathogenicity Avian Influenza H5N1 Season in the Czech Republic. Viruses. 2024; 16(2):221. https://doi.org/10.3390/v16020221
Chicago/Turabian StyleNagy, Alexander, Martina Stará, Lenka Černíková, Eliška Kličková, Ondřej Horák, Lada Hofmannová, and Kamil Sedlák. 2024. "Enzootic Circulation, Massive Gull Mortality and Poultry Outbreaks during the 2022/2023 High-Pathogenicity Avian Influenza H5N1 Season in the Czech Republic" Viruses 16, no. 2: 221. https://doi.org/10.3390/v16020221
APA StyleNagy, A., Stará, M., Černíková, L., Kličková, E., Horák, O., Hofmannová, L., & Sedlák, K. (2024). Enzootic Circulation, Massive Gull Mortality and Poultry Outbreaks during the 2022/2023 High-Pathogenicity Avian Influenza H5N1 Season in the Czech Republic. Viruses, 16(2), 221. https://doi.org/10.3390/v16020221