
Translational Control of Alphavirus–Host Interactions: Implications in Viral Evolution, Tropism and Antiviral Response


{kind=link}
{kind=link}
{kind=link}
Abstract
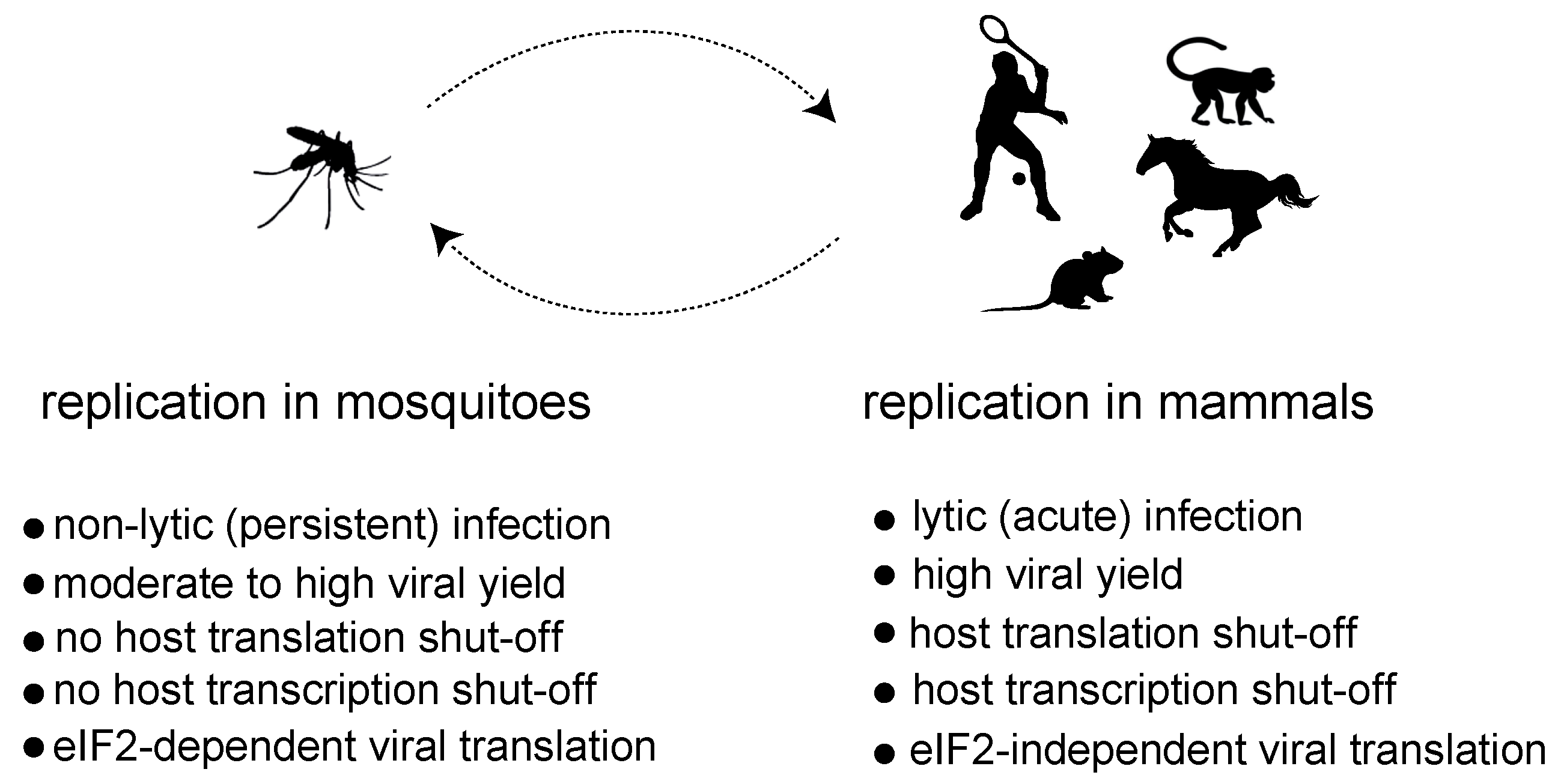
1. Alphavirus Replication, Tropism and Interference with Host Gene Expression
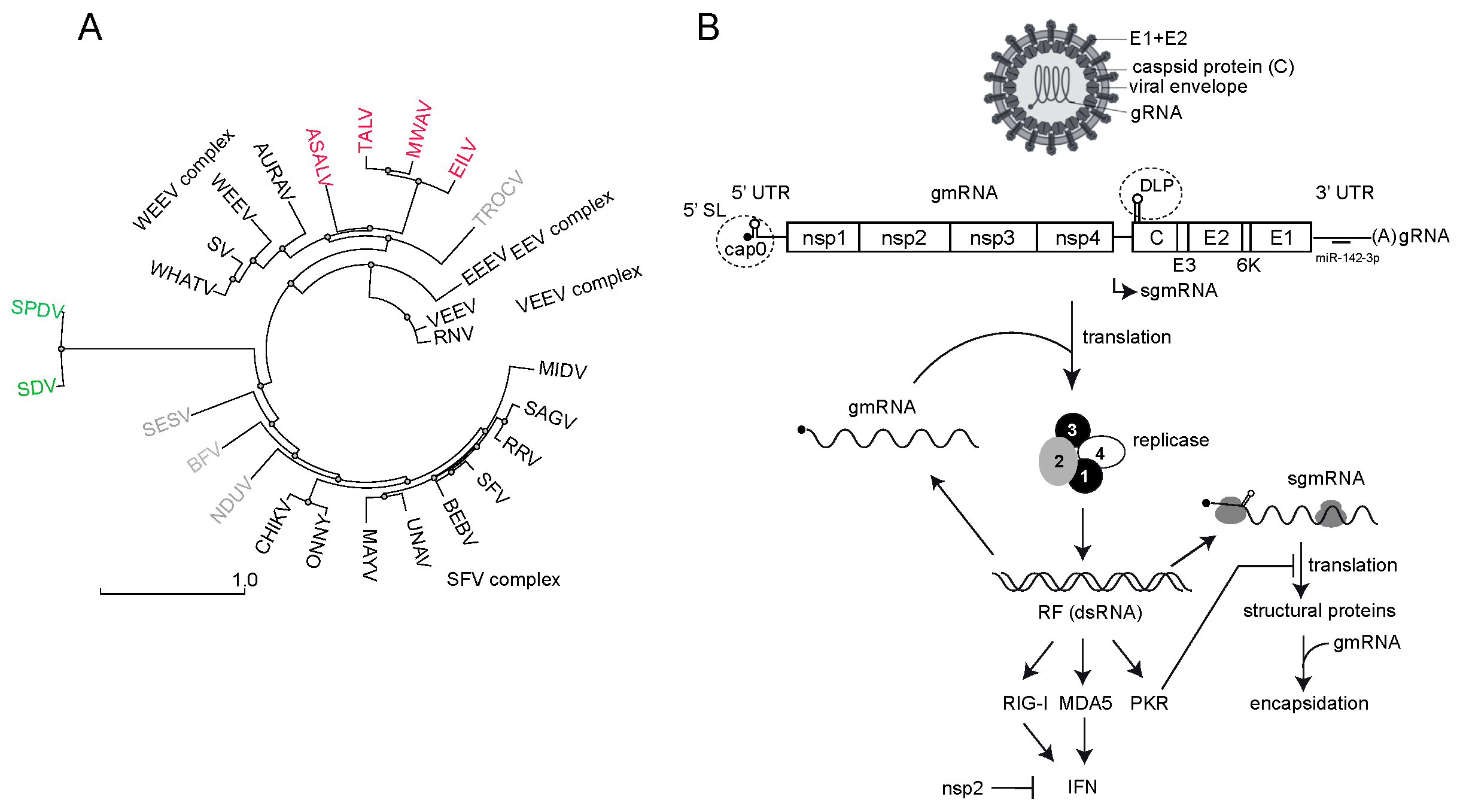
2. Mechanisms of Virus-Induced Host Translation Shutoff
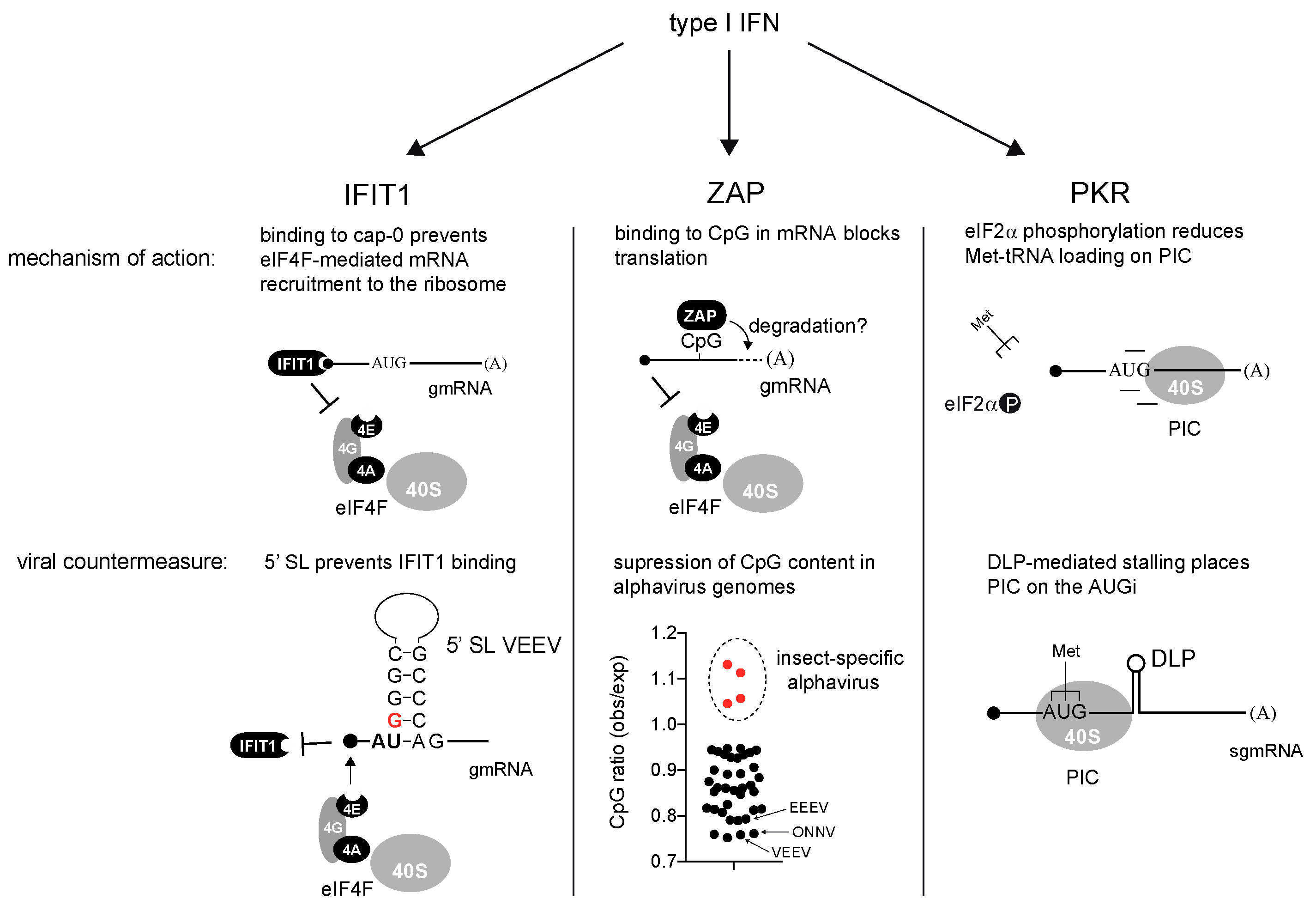
3. Interferon-Induced Genes That Block Translation of Alphaviral mRNAs: Roles of IFIT-1, ZAP and PKR
4. Adaptive Changes in Alphavirus mRNAs to Counteract the Antiviral Effect of IFN
5. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Powers, A.M.; Brault, A.C.; Shirako, Y.; Strauss, E.G.; Kang, W.; Strauss, J.H.; Weaver, S.C. Evolutionary relationships and systematics of the alphaviruses. J. Virol. 2001, 75, 10118–10131. [Google Scholar] [CrossRef]
- Weaver, S.C.; Barrett, A.D.T. Transmission cycles, host range, evolution and emergence of arboviral disease. Nat. Rev. Microbiol. 2004, 2, 789–801. [Google Scholar] [CrossRef]
- Weaver, S.C.; Winegar, R.; Manger, I.D.; Forrester, N.L. Alphaviruses: Population genetics and determinants of emergence. Antivir. Res. 2012, 94, 242–257. [Google Scholar] [CrossRef]
- Tsetsarkin, K.A.; Chen, R.; Sherman, M.B.; Weaver, S.C. Chikungunya virus: Evolution and genetic determinants of emergence. Curr. Opin. Virol. 2011, 1, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Tsetsarkin, K.A.; Chen, R.; Weaver, S.C. Interspecies transmission and chikungunya virus emergence. Curr. Opin. Virol. 2016, 16, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Zacks, M.A.; Paessler, S. Encephalitic alphaviruses. Vet. Microbiol. 2010, 140, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Kafai, N.M.; Diamond, M.S.; Fox, J.M. Distinct Cellular Tropism and Immune Responses to Alphavirus Infection. Annu. Rev. Immunol. 2022, 40, 615–649. [Google Scholar] [CrossRef]
- Strauss, J.H.; Strauss, E.G. The alphaviruses: Gene expression, replication, and evolution. Microbiol. Rev. 1994, 58, 491–562. [Google Scholar] [CrossRef]
- Ahola, T.; McInerney, G.; Merits, A. Alphavirus RNA replication in vertebrate cells. Adv. Virus Res. 2021, 111, 111–156. [Google Scholar] [CrossRef]
- Pettersson, R.F.; Soderlund, H.; Kaariainen, L. The nucleotide sequences of the 5′-terminal T1 oligonucleotides of Semliki-Forest-virus 42-S and 26-S RNAs are different. Eur. J. Biochem. 1980, 105, 435–443. [Google Scholar] [CrossRef]
- Ahola, T.; Kaariainen, L. Reaction in alphavirus mRNA capping: Formation of a covalent complex of nonstructural protein nsP1 with 7-methyl-GMP. Proc. Natl. Acad. Sci. USA 1995, 92, 507–511. [Google Scholar] [CrossRef]
- Ahola, T.; Laakkonen, P.; Vihinen, H.; Kaariainen, L. Critical residues of Semliki Forest virus RNA capping enzyme involved in methyltransferase and guanylyltransferase-like activities. J. Virol. 1997, 71, 392–397. [Google Scholar] [CrossRef]
- Decroly, E.; Ferron, F.; Lescar, J.; Canard, B. Conventional and unconventional mechanisms for capping viral mRNA. Nat. Rev. Microbiol. 2011, 10, 51–65. [Google Scholar] [CrossRef]
- Gorchakov, R.; Frolova, E.; Frolov, I. Inhibition of transcription and translation in Sindbis virus-infected cells. J. Virol. 2005, 79, 9397–9409. [Google Scholar] [CrossRef]
- Gorchakov, R.; Garmashova, N.; Frolova, E.; Frolov, I. Different types of nsP3-containing protein complexes in Sindbis virus-infected cells. J. Virol. 2008, 82, 10088–10101. [Google Scholar] [CrossRef]
- Fros, J.J.; Geertsema, C.; Zouache, K.; Baggen, J.; Domeradzka, N.; van Leeuwen, D.M.; Flipse, J.; Vlak, J.M.; Failloux, A.; Pijlman, G.P. Mosquito Rasputin interacts with chikungunya virus nsP3 and determines the infection rate in Aedes albopictus. Parasit. Vectors 2015, 8, 464. [Google Scholar] [CrossRef]
- Panas, M.D.; Varjak, M.; Lulla, A.; Eng, K.E.; Merits, A.; Karlsson Hedestam, G.B.; McInerney, G.M. Sequestration of G3BP coupled with efficient translation inhibits stress granules in Semliki Forest virus infection. Mol. Biol. Cell 2012, 23, 4701–4712. [Google Scholar] [CrossRef] [PubMed]
- Jayabalan, A.K.; Adivarahan, S.; Koppula, A.; Abraham, R.; Batish, M.; Zenklusen, D.; Griffin, D.E.; Leung, A.K.L. Stress granule formation, disassembly, and composition are regulated by alphavirus ADP-ribosylhydrolase activity. Proc. Natl. Acad. Sci. USA 2021, 118, e2021719118. [Google Scholar] [CrossRef] [PubMed]
- Frolova, E.I.; Palchevska, O.; Dominguez, F.; Frolov, I. Alphavirus-induced transcriptional and translational shutoffs play major roles in blocking the formation of stress granules. J. Virol. 2023, 97, e0097923-23. [Google Scholar] [CrossRef] [PubMed]
- Pietila, M.K.; Hellstrom, K.; Ahola, T. Alphavirus polymerase and RNA replication. Virus Res. 2017, 234, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Lemm, J.A.; Rumenapf, T.; Strauss, E.G.; Strauss, J.H.; Rice, C.M. Polypeptide requirements for assembly of functional Sindbis virus replication complexes: A model for the temporal regulation of minus- and plus-strand RNA synthesis. EMBO J. 1994, 13, 2925–2934. [Google Scholar] [CrossRef]
- Lulla, V.; Karo-Astover, L.; Rausalu, K.; Saul, S.; Merits, A.; Lulla, A. Timeliness of Proteolytic Events Is Prerequisite for Efficient Functioning of the Alphaviral Replicase. J. Virol. 2018, 92, e00151-18. [Google Scholar] [CrossRef]
- Frolova, E.I.; Gorchakov, R.; Pereboeva, L.; Atasheva, S.; Frolov, I. Functional Sindbis virus replicative complexes are formed at the plasma membrane. J. Virol. 2010, 84, 11679–11695. [Google Scholar] [CrossRef]
- Froshauer, S.; Kartenbeck, J.; Helenius, A. Alphavirus RNA replicase is located on the cytoplasmic surface of endosomes and lysosomes. J. Cell Biol. 1988, 107, 2075–2086. [Google Scholar] [CrossRef] [PubMed]
- Laurent, T.; Kumar, P.; Liese, S.; Zare, F.; Jonasson, M.; Carlson, A.; Carlson, L. Architecture of the chikungunya virus replication organelle. eLife 2022, 11, e83042. [Google Scholar] [CrossRef] [PubMed]
- Akhrymuk, I.; Frolov, I.; Frolova, E.I. Both RIG-I and MDA5 detect alphavirus replication in concentration-dependent mode. Virology 2016, 487, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Trobaugh, D.W.; Gardner, C.L.; Sun, C.; Haddow, A.D.; Wang, E.; Chapnik, E.; Mildner, A.; Weaver, S.C.; Ryman, K.D.; Klimstra, W.B. RNA viruses can hijack vertebrate microRNAs to suppress innate immunity. Nature 2014, 506, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Jose, J.; Taylor, A.B.; Kuhn, R.J. Spatial and Temporal Analysis of Alphavirus Replication and Assembly in Mammalian and Mosquito Cells. mBio 2017, 8, e02294-16. [Google Scholar] [CrossRef] [PubMed]
- Gliedman, J.B.; Smith, J.F.; Brown, D.T. Morphogenesis of Sindbis virus in cultured Aedes albopictus cells. J. Virol. 1975, 16, 913–926. [Google Scholar] [CrossRef] [PubMed]
- Davey, M.W.; Dalgarno, L. Semliki Forest virus replication in cultured Aedes albopictus cells: Studies on the establishment of persistence. J. Gen. Virol. 1974, 24, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Rumenapf, T.; Strauss, E.G.; Strauss, J.H. Regulation of Semliki Forest virus RNA replication: A model for the control of alphavirus pathogenesis in invertebrate hosts. Virology 2004, 323, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Ventoso, I. Adaptive changes in alphavirus mRNA translation allowed colonization of vertebrate hosts. J. Virol. 2012, 86, 9484–9494. [Google Scholar] [CrossRef] [PubMed]
- Akhrymuk, I.; Kulemzin, S.V.; Frolova, E.I. Evasion of the innate immune response: The Old World alphavirus nsP2 protein induces rapid degradation of Rpb1, a catalytic subunit of RNA polymerase II. J. Virol. 2012, 86, 7180–7191. [Google Scholar] [CrossRef] [PubMed]
- Stern-Ginossar, N.; Thompson, S.R.; Mathews, M.B.; Mohr, I. Translational Control in Virus-Infected Cells. Cold Spring Harb. Perspect. Biol. 2019, 11, a033001. [Google Scholar] [CrossRef] [PubMed]
- Ventoso, I.; Sanz, M.A.; Molina, S.; Berlanga, J.J.; Carrasco, L.; Esteban, M. Translational resistance of late alphavirus mRNA to eIF2alpha phosphorylation: A strategy to overcome the antiviral effect of protein kinase PKR. Genes Dev. 2006, 20, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Gorchakov, R.; Frolova, E.; Williams, B.R.G.; Rice, C.M.; Frolov, I. PKR-dependent and -independent mechanisms are involved in translational shutoff during Sindbis virus infection. J. Virol. 2004, 78, 8455–8467. [Google Scholar] [CrossRef] [PubMed]
- Meshram, C.D.; Lukash, T.; Phillips, A.T.; Akhrymuk, I.; Frolova, E.I.; Frolov, I. Lack of nsP2-specific nuclear functions attenuates chikungunya virus replication both in vitro and in vivo. Virology 2019, 534, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.; Hauer, D.; McPherson, R.L.; Utt, A.; Kirby, I.T.; Cohen, M.S.; Merits, A.; Leung, A.K.L.; Griffin, D.E. ADP-ribosyl-binding and hydrolase activities of the alphavirus nsP3 macrodomain are critical for initiation of virus replication. Proc. Natl. Acad. Sci. USA 2018, 115, E10457–E10466. [Google Scholar] [CrossRef]
- Dominguez, F.; Palchevska, O.; Frolova, E.I.; Frolov, I. Alphavirus-based replicons demonstrate different interactions with host cells and can be optimized to increase protein expression. J. Virol. 2023, 97, e0122523. [Google Scholar] [CrossRef]
- Bhalla, N.; Sun, C.; Metthew Lam, L.K.; Gardner, C.L.; Ryman, K.D.; Klimstra, W.B. Host translation shutoff mediated by non-structural protein 2 is a critical factor in the antiviral state resistance of Venezuelan equine encephalitis virus. Virology 2016, 496, 147–165. [Google Scholar] [CrossRef]
- Carrasco, L.; Sanz, M.A.; Gonzalez-Almela, E. The Regulation of Translation in Alphavirus-Infected Cells. Viruses 2018, 10, 70. [Google Scholar] [CrossRef]
- Fros, J.J.; Pijlman, G.P. Alphavirus Infection: Host Cell Shut-Off and Inhibition of Antiviral Responses. Viruses 2016, 8, 166. [Google Scholar] [CrossRef]
- McInerney, G.M.; Kedersha, N.L.; Kaufman, R.J.; Anderson, P.; Liljestrom, P. Importance of eIF2alpha phosphorylation and stress granule assembly in alphavirus translation regulation. Mol. Biol. Cell 2005, 16, 3753–3763. [Google Scholar] [CrossRef] [PubMed]
- Toribio, R.; Ventoso, I. Inhibition of host translation by virus infection in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 9837–9842. [Google Scholar] [CrossRef] [PubMed]
- Treffers, E.E.; Tas, A.; Scholte, F.E.M.; de Ru, A.H.; Snijder, E.J.; van Veelen, P.A.; van Hemert, M.J. The alphavirus nonstructural protein 2 NTPase induces a host translational shut-off through phosphorylation of eEF2 via cAMP-PKA-eEF2K signaling. PLoS Pathog. 2023, 19, e1011179. [Google Scholar] [CrossRef] [PubMed]
- Carlberg, U.; Nilsson, A.; Nygard, O. Functional properties of phosphorylated elongation factor 2. Eur. J. Biochem. 1990, 191, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.A.; Garcia-Moreno, M.; Carrasco, L. Inhibition of host protein synthesis by Sindbis virus: Correlation with viral RNA replication and release of nuclear proteins to the cytoplasm. Cell. Microbiol. 2015, 17, 520–541. [Google Scholar] [CrossRef] [PubMed]
- Frolov, I.; Schlesinger, S. Comparison of the effects of Sindbis virus and Sindbis virus replicons on host cell protein synthesis and cytopathogenicity in BHK cells. J. Virol. 1994, 68, 1721–1727. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.A.; Castello, A.; Carrasco, L. Viral translation is coupled to transcription in Sindbis virus-infected cells. J. Virol. 2007, 81, 7061–7068. [Google Scholar] [CrossRef] [PubMed]
- Barnhart, M.D.; Moon, S.L.; Emch, A.W.; Wilusz, C.J.; Wilusz, J. Changes in cellular mRNA stability, splicing, and polyadenylation through HuR protein sequestration by a cytoplasmic RNA virus. Cell Rep. 2013, 5, 909–917. [Google Scholar] [CrossRef]
- van Steeg, H.; Kasperaitis, M.; Voorma, H.O.; Benne, R. Infection of neuroblastoma cells by Semliki Forest virus. The interference of viral capsid protein with the binding of host messenger RNAs into initiation complexes is the cause of the shut-off of host protein synthesis. Eur. J. Biochem. 1984, 138, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Frolov, I.; Akhrymuk, M.; Akhrymuk, I.; Atasheva, S.; Frolova, E.I. Early events in alphavirus replication determine the outcome of infection. J. Virol. 2012, 86, 5055–5066. [Google Scholar] [CrossRef] [PubMed]
- Ryman, K.D.; Klimstra, W.B. Host responses to alphavirus infection. Immunol. Rev. 2008, 225, 27–45. [Google Scholar] [CrossRef] [PubMed]
- Schilte, C.; Couderc, T.; Chretien, F.; Sourisseau, M.; Gangneux, N.; Guivel-Benhassine, F.; Kraxner, A.; Tschopp, J.; Higgs, S.; Michault, A.; et al. Type I IFN controls chikungunya virus via its action on nonhematopoietic cells. J. Exp. Med. 2010, 207, 429–442. [Google Scholar] [CrossRef]
- Grieder, F.B.; Vogel, S.N. Role of interferon and interferon regulatory factors in early protection against Venezuelan equine encephalitis virus infection. Virology 1999, 257, 106–118. [Google Scholar] [CrossRef]
- Ryman, K.D.; Klimstra, W.B.; Nguyen, K.B.; Biron, C.A.; Johnston, R.E. Alpha/beta interferon protects adult mice from fatal Sindbis virus infection and is an important determinant of cell and tissue tropism. J. Virol. 2000, 74, 3366–3378. [Google Scholar] [CrossRef]
- Friedman, R.M.; Fantes, K.H.; Levy, H.B.; Carter, W.B. Interferon action on parental Semliki forest virus ribonucleic acid. J. Virol. 1967, 1, 1168–1173. [Google Scholar] [CrossRef]
- Friedman, R.M. Inhibition of arbovirus protein synthesis by interferon. J. Virol. 1968, 2, 1081–1085. [Google Scholar] [CrossRef]
- Toribio, R.; Diaz-Lopez, I.; Berlanga, J.J.; Molina-Jimenez, F.; Majano, P.; Ventoso, I. Naturally Occurring and Engineered Alphaviruses Sensitive to Double-Stranded-RNA-Activated Protein Kinase Show Restricted Translation in Mammalian Cells, Increased Sensitivity to Interferon, and Marked Oncotropism. J. Virol. 2020, 94, e01630-19. [Google Scholar] [CrossRef]
- Frolova, E.I.; Fayzulin, R.Z.; Cook, S.H.; Griffin, D.E.; Rice, C.M.; Frolov, I. Roles of nonstructural protein nsP2 and Alpha/Beta interferons in determining the outcome of Sindbis virus infection. J. Virol. 2002, 76, 11254–11264. [Google Scholar] [CrossRef]
- Atasheva, S.; Kim, D.Y.; Frolova, E.I.; Frolov, I. Venezuelan equine encephalitis virus variants lacking transcription inhibitory functions demonstrate highly attenuated phenotype. J. Virol. 2015, 89, 71–82. [Google Scholar] [CrossRef]
- Her, Z.; Malleret, B.; Chan, M.; Ong, E.K.S.; Wong, S.; Kwek, D.J.C.; Tolou, H.; Lin, R.T.P.; Tambyah, P.A.; Renia, L.; et al. Active infection of human blood monocytes by Chikungunya virus triggers an innate immune response. J. Immunol. 2010, 184, 5903–5913. [Google Scholar] [CrossRef]
- Sadler, A.J.; Williams, B.R.G. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 2008, 8, 559–568. [Google Scholar] [CrossRef]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472, 481–485. [Google Scholar] [CrossRef]
- Zhang, Y.; Burke, C.W.; Ryman, K.D.; Klimstra, W.B. Identification and characterization of interferon-induced proteins that inhibit alphavirus replication. J. Virol. 2007, 81, 11246–11255. [Google Scholar] [CrossRef]
- McDougal, M.B.; De Maria, A.M.; Ohlson, M.B.; Kumar, A.; Xing, C.; Schoggins, J.W. Interferon inhibits a model RNA virus via a limited set of inducible effector genes. EMBO Rep. 2023, 24, e56901. [Google Scholar] [CrossRef] [PubMed]
- Tesfay, M.Z.; Yin, J.; Gardner, C.L.; Khoretonenko, M.V.; Korneeva, N.L.; Rhoads, R.E.; Ryman, K.D.; Klimstra, W.B. Alpha/beta interferon inhibits cap-dependent translation of viral but not cellular mRNA by a PKR-independent mechanism. J. Virol. 2008, 82, 2620–2630. [Google Scholar] [CrossRef] [PubMed]
- Daffis, S.; Szretter, K.J.; Schriewer, J.; Li, J.; Youn, S.; Errett, J.; Lin, T.; Schneller, S.; Zust, R.; Dong, H.; et al. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 2010, 468, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Hyde, J.L.; Gardner, C.L.; Kimura, T.; White, J.P.; Liu, G.; Trobaugh, D.W.; Huang, C.; Tonelli, M.; Paessler, S.; Takeda, K.; et al. A viral RNA structural element alters host recognition of nonself RNA. Science 2014, 343, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Pichlmair, A.; Lassnig, C.; Eberle, C.; Gorna, M.W.; Baumann, C.L.; Burkard, T.R.; Burckstummer, T.; Stefanovic, A.; Krieger, S.; Bennett, K.L.; et al. IFIT1 is an antiviral protein that recognizes 5′-triphosphate RNA. Nat. Immunol. 2011, 12, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Abbas, Y.M.; Pichlmair, A.; Gorna, M.W.; Superti-Furga, G.; Nagar, B. Structural basis for viral 5′-PPP-RNA recognition by human IFIT proteins. Nature 2013, 494, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Sweeney, T.R.; Skabkin, M.A.; Skabkina, O.V.; Hellen, C.U.T.; Pestova, T.V. Inhibition of translation by IFIT family members is determined by their ability to interact selectively with the 5′-terminal regions of cap0-, cap1- and 5′ppp-mRNAs. Nucleic Acids Res. 2014, 42, 3228–3245. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.; VanBlargan, L.A.; Xu, W.; White, J.P.; Shan, C.; Shi, P.; Zhang, R.; Adhikari, J.; Gross, M.L.; Leung, D.W.; et al. Human IFIT3 Modulates IFIT1 RNA Binding Specificity and Protein Stability. Immunity 2018, 48, 487–499.e5. [Google Scholar] [CrossRef] [PubMed]
- Fleith, R.C.; Mears, H.V.; Leong, X.Y.; Sanford, T.J.; Emmott, E.; Graham, S.C.; Mansur, D.S.; Sweeney, T.R. IFIT3 and IFIT2/3 promote IFIT1-mediated translation inhibition by enhancing binding to non-self RNA. Nucleic Acids Res. 2018, 46, 5269–5285. [Google Scholar] [CrossRef] [PubMed]
- Reynaud, J.M.; Kim, D.Y.; Atasheva, S.; Rasalouskaya, A.; White, J.P.; Diamond, M.S.; Weaver, S.C.; Frolova, E.I.; Frolov, I. IFIT1 Differentially Interferes with Translation and Replication of Alphavirus Genomes and Promotes Induction of Type I Interferon. PLoS Pathog. 2015, 11, e1004863. [Google Scholar] [CrossRef] [PubMed]
- Bick, M.J.; Carroll, J.N.; Gao, G.; Goff, S.P.; Rice, C.M.; MacDonald, M.R. Expression of the zinc-finger antiviral protein inhibits alphavirus replication. J. Virol. 2003, 77, 11555–11562. [Google Scholar] [CrossRef]
- Ficarelli, M.; Neil, S.J.D.; Swanson, C.M. Targeted Restriction of Viral Gene Expression and Replication by the ZAP Antiviral System. Annu. Rev. Virol. 2021, 8, 265–283. [Google Scholar] [CrossRef]
- Goncalves-Carneiro, D.; Takata, M.A.; Ong, H.; Shilton, A.; Bieniasz, P.D. Origin and evolution of the zinc finger antiviral protein. PLoS Pathog. 2021, 17, e1009545. [Google Scholar] [CrossRef]
- Gao, G.; Guo, X.; Goff, S.P. Inhibition of retroviral RNA production by ZAP, a CCCH-type zinc finger protein. Science 2002, 297, 1703–1706. [Google Scholar] [CrossRef]
- Guo, X.; Carroll, J.N.; Macdonald, M.R.; Goff, S.P.; Gao, G. The zinc finger antiviral protein directly binds to specific viral mRNAs through the CCCH zinc finger motifs. J. Virol. 2004, 78, 12781–12787. [Google Scholar] [CrossRef]
- Schwerk, J.; Soveg, F.W.; Ryan, A.P.; Thomas, K.R.; Hatfield, L.D.; Ozarkar, S.; Forero, A.; Kell, A.M.; Roby, J.A.; So, L.; et al. RNA-binding protein isoforms ZAP-S and ZAP-L have distinct antiviral and immune resolution functions. Nat. Immunol. 2019, 20, 1610–1620. [Google Scholar] [CrossRef]
- Li, M.M.H.; Aguilar, E.G.; Michailidis, E.; Pabon, J.; Park, P.; Wu, X.; de Jong, Y.P.; Schneider, W.M.; Molina, H.; Rice, C.M.; et al. Characterization of Novel Splice Variants of Zinc Finger Antiviral Protein (ZAP). J. Virol. 2019, 93, e00715-19. [Google Scholar] [CrossRef]
- Kerns, J.A.; Emerman, M.; Malik, H.S. Positive selection and increased antiviral activity associated with the PARP-containing isoform of human zinc-finger antiviral protein. PLoS Genet. 2008, 4, e21. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, X.; Goff, S.P.; Gao, G. Translational repression precedes and is required for ZAP-mediated mRNA decay. EMBO J. 2012, 31, 4236–4246. [Google Scholar] [CrossRef]
- Takata, M.A.; Goncalves-Carneiro, D.; Zang, T.M.; Soll, S.J.; York, A.; Blanco-Melo, D.; Bieniasz, P.D. CG dinucleotide suppression enables antiviral defence targeting non-self RNA. Nature 2017, 550, 124–127. [Google Scholar] [CrossRef]
- Luo, X.; Wang, X.; Gao, Y.; Zhu, J.; Liu, S.; Gao, G.; Gao, P. Molecular Mechanism of RNA Recognition by Zinc-Finger Antiviral Protein. Cell Rep. 2020, 30, 46–52.e4. [Google Scholar] [CrossRef]
- Meagher, J.L.; Takata, M.; Goncalves-Carneiro, D.; Keane, S.C.; Rebendenne, A.; Ong, H.; Orr, V.K.; MacDonald, M.R.; Stuckey, J.A.; Bieniasz, P.D.; et al. Structure of the zinc-finger antiviral protein in complex with RNA reveals a mechanism for selective targeting of CG-rich viral sequences. Proc. Natl. Acad. Sci. USA 2019, 116, 24303–24309. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Xia, W.; Baillie, J.K.; McKinnon, K. Modelling mutational and selection pressures on dinucleotides in eukaryotic phyla—Selection against CpG and UpA in cytoplasmically expressed RNA and in RNA viruses. BMC Genom. 2013, 14, 610. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.P.; Aldana, K.S.; Yang, E.; Yao, Z.; Li, M.M.H. Alphavirus Evasion of Zinc Finger Antiviral Protein (ZAP) Correlates with CpG Suppression in a Specific Viral nsP2 Gene Sequence. Viruses 2023, 15, 830. [Google Scholar] [CrossRef] [PubMed]
- Goonawardane, N.; Nguyen, D.; Simmonds, P. Association of Zinc Finger Antiviral Protein Binding to Viral Genomic RNA with Attenuation of Replication of Echovirus 7. mSphere 2021, 6, e01138-20. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.; Nguyen, L.P.; Wisherop, C.A.; Kan, R.L.; Li, M.M.H. The Role of ZAP and TRIM25 RNA Binding in Restricting Viral Translation. Front. Cell. Infect. Microbiol. 2022, 12, 886929. [Google Scholar] [CrossRef]
- Kozaki, T.; Takahama, M.; Misawa, T.; Matsuura, Y.; Akira, S.; Saitoh, T. Role of zinc-finger anti-viral protein in host defense against Sindbis virus. Int. Immunol. 2015, 27, 357–364. [Google Scholar] [CrossRef]
- Li, M.M.H.; Lau, Z.; Cheung, P.; Aguilar, E.G.; Schneider, W.M.; Bozzacco, L.; Molina, H.; Buehler, E.; Takaoka, A.; Rice, C.M.; et al. TRIM25 Enhances the Antiviral Action of Zinc-Finger Antiviral Protein (ZAP). PLoS Pathog. 2017, 13, e1006145. [Google Scholar] [CrossRef]
- Roberts, W.K.; Hovanessian, A.; Brown, R.E.; Clemens, M.J.; Kerr, I.M. Interferon-mediated protein kinase and low-molecular-weight inhibitor of protein synthesis. Nature 1976, 264, 477–480. [Google Scholar] [CrossRef]
- Meurs, E.; Chong, K.; Galabru, J.; Thomas, N.S.; Kerr, I.M.; Williams, B.R.; Hovanessian, A.G. Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell 1990, 62, 379–390. [Google Scholar] [CrossRef]
- Garcia, M.A.; Gil, J.; Ventoso, I.; Guerra, S.; Domingo, E.; Rivas, C.; Esteban, M. Impact of protein kinase PKR in cell biology: From antiviral to antiproliferative action. Microbiol. Mol. Biol. Rev. 2006, 70, 1032–1060. [Google Scholar] [CrossRef] [PubMed]
- Chaumont, L.; Collet, B.; Boudinot, P. Double-stranded RNA-dependent protein kinase (PKR) in antiviral defence in fish and mammals. Dev. Comp. Immunol. 2023, 145, 104732. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.L.; Reis, L.F.; Pavlovic, J.; Aguzzi, A.; Schafer, R.; Kumar, A.; Williams, B.R.; Aguet, M.; Weissmann, C. Deficient signaling in mice devoid of double-stranded RNA-dependent protein kinase. EMBO J. 1995, 14, 6095–6106. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, S.; Roberts, P.C.; Brown, L.E.; Truong, H.; Pattnaik, A.K.; Archer, D.R.; Barber, G.N. Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity 2000, 13, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Gordiyenko, Y.; Llacer, J.L.; Ramakrishnan, V. Structural basis for the inhibition of translation through eIF2alpha phosphorylation. Nat. Commun. 2019, 10, 2640. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, T.; Pavitt, G.D.; Zhang, F.; Dever, T.E.; Hinnebusch, A.G. Tight binding of the phosphorylated alpha subunit of initiation factor 2 (eIF2alpha) to the regulatory subunits of guanine nucleotide exchange factor eIF2B is required for inhibition of translation initiation. Mol. Cell. Biol. 2001, 21, 5018–5030. [Google Scholar] [CrossRef] [PubMed]
- Cesaro, T.; Michiels, T. Inhibition of PKR by Viruses. Front. Microbiol. 2021, 12, 757238. [Google Scholar] [CrossRef]
- Garcia-Sastre, A. Ten Strategies of Interferon Evasion by Viruses. Cell Host Microbe 2017, 22, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, P.V.; Adams, A.P.; Wang, E.; Kang, W.; Carrara, A.; Anishchenko, M.; Frolov, I.; Weaver, S.C. Structural and nonstructural protein genome regions of eastern equine encephalitis virus are determinants of interferon sensitivity and murine virulence. J. Virol. 2008, 82, 4920–4930. [Google Scholar] [CrossRef] [PubMed]
- Torii, S.; Orba, Y.; Hang’ombe, B.M.; Mweene, A.S.; Wada, Y.; Anindita, P.D.; Phongphaew, W.; Qiu, Y.; Kajihara, M.; Mori-Kajihara, A.; et al. Discovery of Mwinilunga alphavirus: A novel alphavirus in Culex mosquitoes in Zambia. Virus Res. 2018, 250, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Hermanns, K.; Marklewitz, M.; Zirkel, F.; Overheul, G.J.; Page, R.A.; Loaiza, J.R.; Drosten, C.; van Rij, R.P.; Junglen, S. Agua Salud alphavirus defines a novel lineage of insect-specific alphaviruses discovered in the New World. J. Gen. Virol. 2020, 101, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Nasar, F.; Palacios, G.; Gorchakov, R.V.; Guzman, H.; Da Rosa, A.P.T.; Savji, N.; Popov, V.L.; Sherman, M.B.; Lipkin, W.I.; Tesh, R.B.; et al. Eilat virus, a unique alphavirus with host range restricted to insects by RNA replication. Proc. Natl. Acad. Sci. USA 2012, 109, 14622–14627. [Google Scholar] [CrossRef]
- Hermanns, K.; Zirkel, F.; Kopp, A.; Marklewitz, M.; Rwego, I.B.; Estrada, A.; Gillespie, T.R.; Drosten, C.; Junglen, S. Discovery of a novel alphavirus related to Eilat virus. J. Gen. Virol. 2017, 98, 43–49. [Google Scholar] [CrossRef]
- Odon, V.; Fiddaman, S.R.; Smith, A.L.; Simmonds, P. Comparison of CpG- and UpA-mediated restriction of RNA virus replication in mammalian and avian cells and investigation of potential ZAP-mediated shaping of host transcriptome compositions. RNA 2022, 28, 1089–1109. [Google Scholar] [CrossRef]
- Toribio, R.; Diaz-Lopez, I.; Boskovic, J.; Ventoso, I. An RNA trapping mechanism in Alphavirus mRNA promotes ribosome stalling and translation initiation. Nucleic Acids Res. 2016, 44, 4368–4380. [Google Scholar] [CrossRef]
- Frolov, I.; Schlesinger, S. Translation of Sindbis virus mRNA: Effects of sequences downstream of the initiating codon. J. Virol. 1994, 68, 8111–8117. [Google Scholar] [CrossRef]
- Rathore, A.P.S.; Ng, M.; Vasudevan, S.G. Differential unfolded protein response during Chikungunya and Sindbis virus infection: CHIKV nsP4 suppresses eIF2alpha phosphorylation. Virol. J. 2013, 10, 36. [Google Scholar] [CrossRef]
- Garmashova, N.; Gorchakov, R.; Volkova, E.; Paessler, S.; Frolova, E.; Frolov, I. The Old World and New World alphaviruses use different virus-specific proteins for induction of transcriptional shutoff. J. Virol. 2007, 81, 2472–2484. [Google Scholar] [CrossRef]
- Garmashova, N.; Gorchakov, R.; Frolova, E.; Frolov, I. Sindbis virus nonstructural protein nsP2 is cytotoxic and inhibits cellular transcription. J. Virol. 2006, 80, 5686–5696. [Google Scholar] [CrossRef]
- Atasheva, S.; Garmashova, N.; Frolov, I.; Frolova, E. Venezuelan equine encephalitis virus capsid protein inhibits nuclear import in Mammalian but not in mosquito cells. J. Virol. 2008, 82, 4028–4041. [Google Scholar] [CrossRef] [PubMed]
- Akhrymuk, I.; Lukash, T.; Frolov, I.; Frolova, E.I. Novel Mutations in nsP2 Abolish Chikungunya Virus-Induced Transcriptional Shutoff and Make the Virus Less Cytopathic without Affecting Its Replication Rates. J. Virol. 2019, 93, e02062-18. [Google Scholar] [CrossRef] [PubMed]
- Atasheva, S.; Fish, A.; Fornerod, M.; Frolova, E.I. Venezuelan equine Encephalitis virus capsid protein forms a tetrameric complex with CRM1 and importin alpha/beta that obstructs nuclear pore complex function. J. Virol. 2010, 84, 4158–4171. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ventoso, I.; Berlanga, J.J.; Toribio, R.; Díaz-López, I. Translational Control of Alphavirus–Host Interactions: Implications in Viral Evolution, Tropism and Antiviral Response. Viruses 2024, 16, 205. https://doi.org/10.3390/v16020205
Ventoso I, Berlanga JJ, Toribio R, Díaz-López I. Translational Control of Alphavirus–Host Interactions: Implications in Viral Evolution, Tropism and Antiviral Response. Viruses. 2024; 16(2):205. https://doi.org/10.3390/v16020205
Chicago/Turabian StyleVentoso, Iván, Juan José Berlanga, René Toribio, and Irene Díaz-López. 2024. "Translational Control of Alphavirus–Host Interactions: Implications in Viral Evolution, Tropism and Antiviral Response" Viruses 16, no. 2: 205. https://doi.org/10.3390/v16020205
APA StyleVentoso, I., Berlanga, J. J., Toribio, R., & Díaz-López, I. (2024). Translational Control of Alphavirus–Host Interactions: Implications in Viral Evolution, Tropism and Antiviral Response. Viruses, 16(2), 205. https://doi.org/10.3390/v16020205