
Enhanced Transcription of Human Endogenous Retroviruses and TRIM28 Downregulation in Patients with Inflammatory Bowel Disease


 , ,
, ,  ,
,
Abstract
1. Introduction
2. Material and Methods
2.1. Study Populations
2.2. Sample Storage
2.3. Total RNA Extraction
2.4. Reverse Transcription
2.5. Transcription Levels of pol Genes of HERV-H, -K, -W; env Genes of SYN1, SYN2, and HERV-W; as Well as TRIM8/SETDB1 by a Real-Time PCR Assay
2.6. Statistical Analysis
3. Results
3.1. Study Population
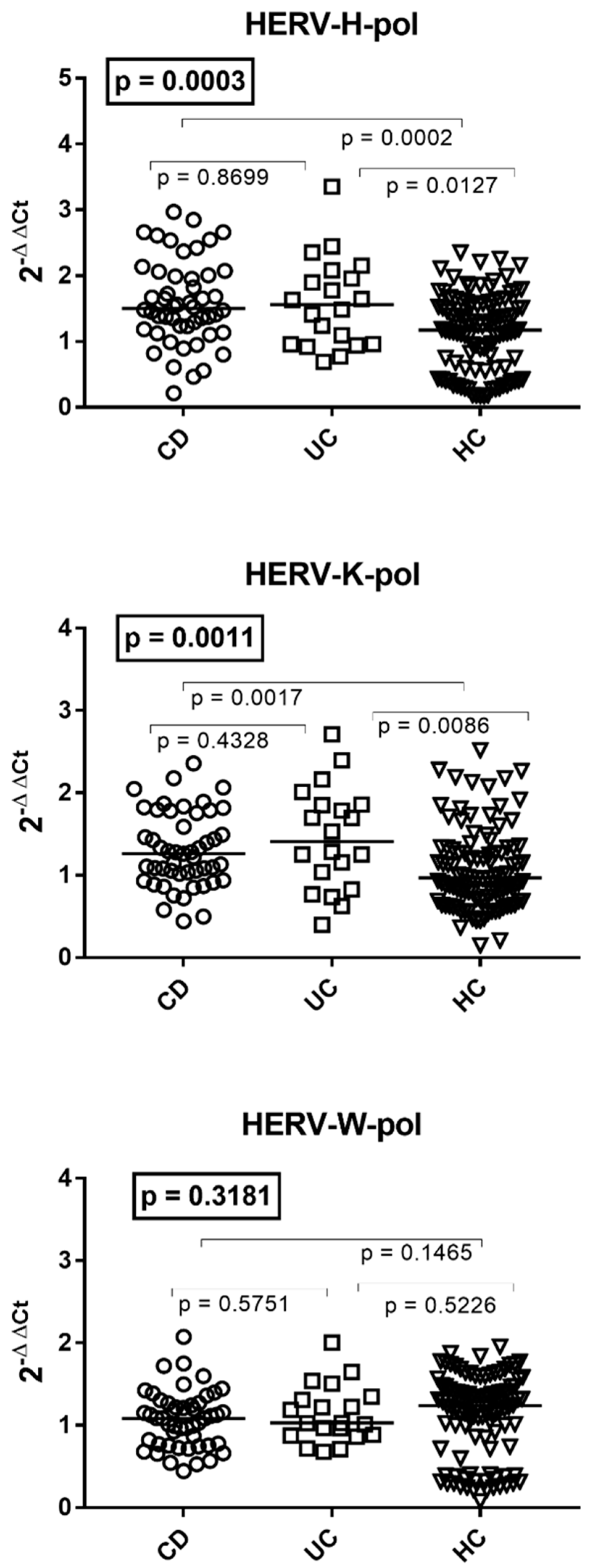
3.2. Transcription Levels of HERV-H-pol, HERV-K-pol, and HERV-W-pol in the Whole Blood of Patients with Crohn’s Disease, Ulcerative Colitis, and HC
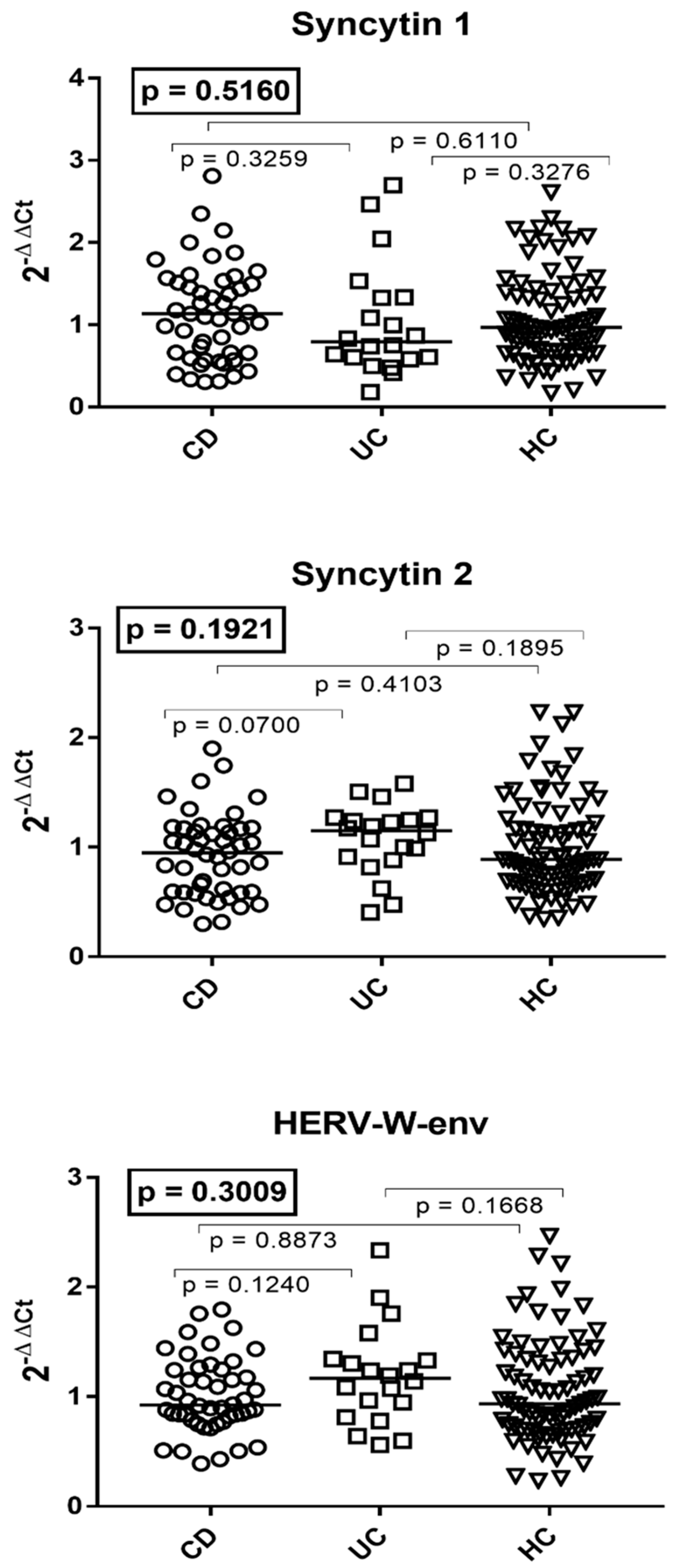
3.3. Transcription Levels of the env Genes of Syncytin 1, Syncytin 2, and HERV-W in the Whole Blood of Patients with Crohn’s Disease, Ulcerative Colitis, and HC
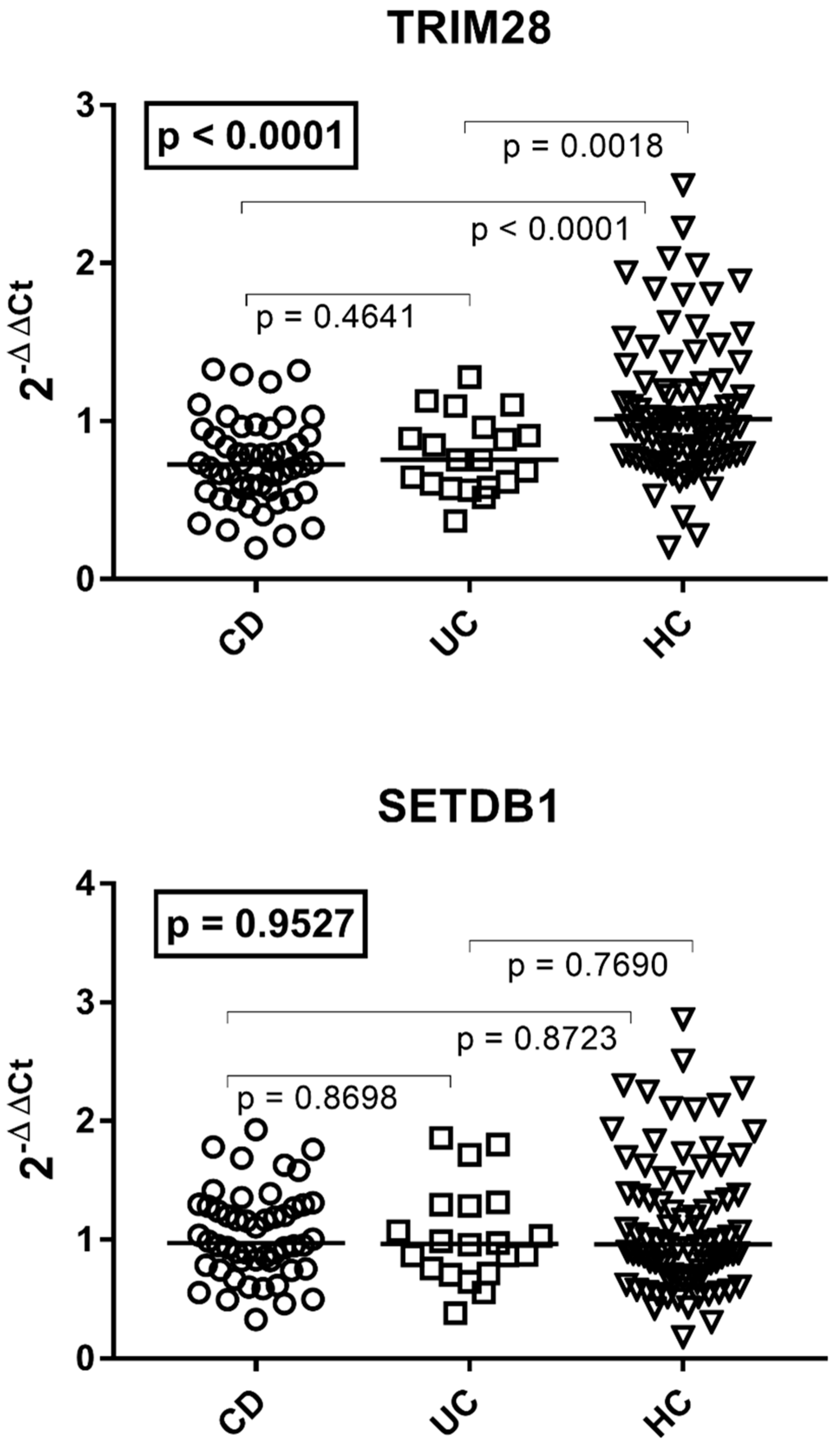
3.4. Transcription Levels of TRIM28 and SETDB1 in Patients with Crohn’s Disease, Ulcerative Colitis, and HC
3.5. Expressions of HERVs, TRIM28, and SETDB1 in IBD Patients According to Disease Activity
3.6. Transcription Levels of HERVs, TRIM28, and SETDB1 in IBD Patients According to Mesalazine Treatment
3.7. Transcription Levels of HERVs, TRIM28, and SETDB1 in IBD Patients According to Steroid Treatment
3.8. Expressions of HERVs, TRIM28, and SETDB1 in IBD Patients with and without Anti-Tumor Necrosis Factor (TNF) Treatment
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Baumgart, D.C.; Carding, S.R. Inflammatory Bowel Disease: Cause and Immunobiology. Lancet 2007, 369, 1627–1640. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F. Strategies for Targeting Cytokines in Inflammatory Bowel Disease. Nat. Rev. Immunol. 2024, 24, 559–576. [Google Scholar] [CrossRef] [PubMed]
- Jarmakiewicz-Czaja, S.; Zielińska, M.; Sokal, A.; Filip, R. Genetic and Epigenetic Etiology of Inflammatory Bowel Disease: An Update. Genes 2022, 13, 2388. [Google Scholar] [CrossRef] [PubMed]
- Kalla, R.; Adams, A.T.; Nowak, J.K.; Bergemalm, D.; Vatn, S.; Ventham, N.T.; Kennedy, N.A.; Ricanek, P.; Lindstrom, J.; IBD-Character Consortium; et al. Analysis of Systemic Epigenetic Alterations in Inflammatory Bowel Disease: Defining Geographical, Genetic and Immune-Inflammatory Influences on the Circulating Methylome. J. Crohns Colitis 2023, 17, 170–184. [Google Scholar] [CrossRef] [PubMed]
- Stankey, C.T.; Bourges, C.; Haag, L.M.; Turner-Stokes, T.; Piedade, A.P.; Palmer-Jones, C.; Papa, I.; Silva Dos Santos, M.; Zhang, Q.; Cameron, A.J.; et al. A Disease-Associated Gene Desert Directs Macrophage Inflammation through ETS2. Nature 2024, 630, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Noble, A.J.; Nowak, J.K.; Adams, A.T.; Uhlig, H.H.; Satsangi, J. Defining Interactions Between the Genome, Epigenome, and the Environment in Inflammatory Bowel Disease: Progress and Prospects. Gastroenterology 2023, 165, 44–60.e2. [Google Scholar] [CrossRef]
- Ventham, N.T.; Kennedy, N.A.; Kalla, R.; Adams, A.T.; Noble, A.; Ennis, H.; TOPPIC Study Group; IBD-BIOM Consortium; Mowat, C.; Dunlop, M.G.; et al. Genome-Wide Methylation Profiling in 229 Patients With Crohn’s Disease Requiring Intestinal Resection: Epigenetic Analysis of the Trial of Prevention of Post-Operative Crohn’s Disease (TOPPIC). Cell Mol. Gastroenterol. Hepatol. 2023, 16, 431–450. [Google Scholar] [CrossRef]
- Bastida, G.; Mínguez, A.; Nos, P.; Moret-Tatay, I. Immunoepigenetic Regulation of Inflammatory Bowel Disease: Current Insights into Novel Epigenetic Modulations of the Systemic Immune Response. Genes 2023, 14, 554. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Groer, M.; Dutra, S.V.O.; Sarkar, A.; McSkimming, D.I. Gut Microbiota and Immune System Interactions. Microorganisms 2020, 8, 1587. [Google Scholar] [CrossRef]
- Quaglio, A.E.V.; Grillo, T.G.; De Oliveira, E.C.S.; Di Stasi, L.C.; Sassaki, L.Y. Gut Microbiota, Inflammatory Bowel Disease and Colorectal Cancer. World J. Gastroenterol. 2022, 28, 4053–4060. [Google Scholar] [CrossRef]
- Liang, H.; Zhang, L.; Hoden, B.; Qu, B.; Derubeis, D.; Song, X.; Zhang, D. Delineating the Role of Toll-Like Receptors in Inflammatory Bowel Disease. Methods Mol. Biol. 2023, 2700, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.E. Origins and Evolutionary Consequences of Ancient Endogenous Retroviruses. Nat. Rev. Microbiol. 2019, 17, 355–370. [Google Scholar] [CrossRef]
- Blond, J.L.; Lavillette, D.; Cheynet, V.; Bouton, O.; Oriol, G.; Chapel-Fernandes, S.; Mandrand, B.; Mallet, F.; Cosset, F.L. An Envelope Glycoprotein of the Human Endogenous Retrovirus HERV-W Is Expressed in the Human Placenta and Fuses Cells Expressing the Type D Mammalian Retrovirus Receptor. J. Virol. 2000, 74, 3321–3329. [Google Scholar] [CrossRef] [PubMed]
- Blaise, S.; de Parseval, N.; Bénit, L.; Heidmann, T. Genomewide Screening for Fusogenic Human Endogenous Retrovirus Envelopes Identifies Syncytin 2, a Gene Conserved on Primate Evolution. Proc. Natl. Acad. Sci. USA 2003, 100, 13013–13018. [Google Scholar] [CrossRef]
- Garcia-Montojo, M.; Rodriguez-Martin, E.; Ramos-Mozo, P.; Ortega-Madueño, I.; Dominguez-Mozo, M.I.; Arias-Leal, A.; García-Martínez, M.Á.; Casanova, I.; Galan, V.; Arroyo, R.; et al. Syncytin-1/HERV-W Envelope Is an Early Activation Marker of Leukocytes and Is Upregulated in Multiple Sclerosis Patients. Eur. J. Immunol. 2020, 50, 685–694. [Google Scholar] [CrossRef]
- Isbel, L.; Whitelaw, E. Endogenous Retroviruses in Mammals: An Emerging Picture of How ERVs Modify Expression of Adjacent Genes. Bioessays 2012, 34, 734–738. [Google Scholar] [CrossRef] [PubMed]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory Evolution of Innate Immunity through Co-Option of Endogenous Retroviruses. Science 2016, 351, 1083–1087. [Google Scholar] [CrossRef]
- Miyake, K.; Shibata, T.; Ohto, U.; Shimizu, T.; Saitoh, S.-I.; Fukui, R.; Murakami, Y. Mechanisms Controlling Nucleic Acid-Sensing Toll-like Receptors. Int. Immunol. 2018, 30, 43–51. [Google Scholar] [CrossRef]
- Dembny, P.; Newman, A.G.; Singh, M.; Hinz, M.; Szczepek, M.; Krüger, C.; Adalbert, R.; Dzaye, O.; Trimbuch, T.; Wallach, T.; et al. Human Endogenous Retrovirus HERV-K(HML-2) RNA Causes Neurodegeneration through Toll-like Receptors. JCI Insight 2020, 5, e131093. [Google Scholar] [CrossRef]
- Volkman, H.E.; Stetson, D.B. The Enemy within: Endogenous Retroelements and Autoimmune Disease. Nat. Immunol. 2014, 15, 415–422. [Google Scholar] [CrossRef]
- Perron, H.; Dougier-Reynaud, H.-L.; Lomparski, C.; Popa, I.; Firouzi, R.; Bertrand, J.-B.; Marusic, S.; Portoukalian, J.; Jouvin-Marche, E.; Villiers, C.L.; et al. Human Endogenous Retrovirus Protein Activates Innate Immunity and Promotes Experimental Allergic Encephalomyelitis in Mice. PLoS ONE 2013, 8, e80128. [Google Scholar] [CrossRef] [PubMed]
- Tovo, P.-A.; Rabbone, I.; Tinti, D.; Galliano, I.; Trada, M.; Daprà, V.; Cerutti, F.; Bergallo, M. Enhanced Expression of Human Endogenous Retroviruses in New-Onset Type 1 Diabetes: Potential Pathogenetic and Therapeutic Implications. Autoimmunity 2020, 53, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Tovo, P.-A.; Opramolla, A.; Pizzol, A.; Calosso, G.; Daprà, V.; Galliano, I.; Calvi, C.; Pinon, M.; Cisarò, F.; Rigazio, C.; et al. Overexpression of Endogenous Retroviruses in Children with Celiac Disease. Eur. J. Pediatr. 2021, 180, 2429–2434. [Google Scholar] [CrossRef]
- Tovo, P.-A.; Galliano, I.; Parodi, E.; Calvi, C.; Gambarino, S.; Licciardi, F.; Dini, M.; Montanari, P.; Branca, M.; Ramenghi, U.; et al. Children with Chronic Immune Thrombocytopenia Exhibit High Expression of Human Endogenous Retroviruses TRIM28 and SETDB1. Genes 2023, 14, 1569. [Google Scholar] [CrossRef]
- Lima-Junior, D.S.; Krishnamurthy, S.R.; Bouladoux, N.; Collins, N.; Han, S.-J.; Chen, E.Y.; Constantinides, M.G.; Link, V.M.; Lim, A.I.; Enamorado, M.; et al. Endogenous Retroviruses Promote Homeostatic and Inflammatory Responses to the Microbiota. Cell 2021, 184, 3794–3811.e19. [Google Scholar] [CrossRef]
- Friedman, J.R.; Fredericks, W.J.; Jensen, D.E.; Speicher, D.W.; Huang, X.P.; Neilson, E.G.; Rauscher, F.J. KAP-1, a Novel Corepressor for the Highly Conserved KRAB Repression Domain. Genes Dev. 1996, 10, 2067–2078. [Google Scholar] [CrossRef]
- Schultz, D.C.; Ayyanathan, K.; Negorev, D.; Maul, G.G.; Rauscher, F.J. SETDB1: A Novel KAP-1-Associated Histone H3, Lysine 9-Specific Methyltransferase That Contributes to HP1-Mediated Silencing of Euchromatic Genes by KRAB Zinc-Finger Proteins. Genes Dev. 2002, 16, 919–932. [Google Scholar] [CrossRef]
- Wang, R.; Li, H.; Wu, J.; Cai, Z.-Y.; Li, B.; Ni, H.; Qiu, X.; Chen, H.; Liu, W.; Yang, Z.-H.; et al. Gut Stem Cell Necroptosis by Genome Instability Triggers Bowel Inflammation. Nature 2020, 580, 386–390. [Google Scholar] [CrossRef]
- Južnić, L.; Peuker, K.; Strigli, A.; Brosch, M.; Herrmann, A.; Häsler, R.; Koch, M.; Matthiesen, L.; Zeissig, Y.; Löscher, B.-S.; et al. SETDB1 Is Required for Intestinal Epithelial Differentiation and the Prevention of Intestinal Inflammation. Gut 2021, 70, 485–498. [Google Scholar] [CrossRef]
- Takikita, S.; Muro, R.; Takai, T.; Otsubo, T.; Kawamura, Y.I.; Dohi, T.; Oda, H.; Kitajima, M.; Oshima, K.; Hattori, M.; et al. A Histone Methyltransferase ESET Is Critical for T Cell Development. J. Immunol. 2016, 197, 2269–2279. [Google Scholar] [CrossRef]
- Matsui, T.; Leung, D.; Miyashita, H.; Maksakova, I.A.; Miyachi, H.; Kimura, H.; Tachibana, M.; Lorincz, M.C.; Shinkai, Y. Proviral Silencing in Embryonic Stem Cells Requires the Histone Methyltransferase ESET. Nature 2010, 464, 927–931. [Google Scholar] [CrossRef] [PubMed]
- Rowe, H.M.; Friedli, M.; Offner, S.; Verp, S.; Mesnard, D.; Marquis, J.; Aktas, T.; Trono, D. De Novo DNA Methylation of Endogenous Retroviruses Is Shaped by KRAB-ZFPs/KAP1 and ESET. Development 2013, 140, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Turelli, P.; Castro-Diaz, N.; Marzetta, F.; Kapopoulou, A.; Raclot, C.; Duc, J.; Tieng, V.; Quenneville, S.; Trono, D. Interplay of TRIM28 and DNA Methylation in Controlling Human Endogenous Retroelements. Genome Res. 2014, 24, 1260–1270. [Google Scholar] [CrossRef] [PubMed]
- Groner, A.C.; Meylan, S.; Ciuffi, A.; Zangger, N.; Ambrosini, G.; Dénervaud, N.; Bucher, P.; Trono, D. KRAB-Zinc Finger Proteins and KAP1 Can Mediate Long-Range Transcriptional Repression through Heterochromatin Spreading. PLoS Genet. 2010, 6, e1000869. [Google Scholar] [CrossRef]
- Krischuns, T.; Günl, F.; Henschel, L.; Binder, M.; Willemsen, J.; Schloer, S.; Rescher, U.; Gerlt, V.; Zimmer, G.; Nordhoff, C.; et al. Phosphorylation of TRIM28 Enhances the Expression of IFN-β and Proinflammatory Cytokines During HPAIV Infection of Human Lung Epithelial Cells. Front. Immunol. 2018, 9, 2229. [Google Scholar] [CrossRef]
- Gehrmann, U.; Burbage, M.; Zueva, E.; Goudot, C.; Esnault, C.; Ye, M.; Carpier, J.-M.; Burgdorf, N.; Hoyler, T.; Suarez, G.; et al. Critical Role for TRIM28 and HP1β/γ in the Epigenetic Control of T Cell Metabolic Reprograming and Effector Differentiation. Proc. Natl. Acad. Sci. USA 2019, 116, 25839–25849. [Google Scholar] [CrossRef]
- Czerwinska, P.; Jaworska, A.M.; Wlodarczyk, N.A.; Mackiewicz, A.A. Melanoma Stem Cell-Like Phenotype and Significant Suppression of Immune Response within a Tumor Are Regulated by TRIM28 Protein. Cancers 2020, 12, 2998. [Google Scholar] [CrossRef]
- Klag, T.; Courth, L.; Ostaff, M.J.; Ott, G.; Stange, E.F.; Malek, N.P.; Seifarth, W.; Wehkamp, J. Human Endogenous Retroviruses: Residues of Ancient Times Are Differentially Expressed in Crohn’s Disease. Inflamm. Intestig. Dis. 2019, 3, 125–137. [Google Scholar] [CrossRef]
- Grandi, N.; Tramontano, E. Human Endogenous Retroviruses Are Ancient Acquired Elements Still Shaping Innate Immune Responses. Front. Immunol. 2018, 9, 2039. [Google Scholar] [CrossRef]
- Rolland, A.; Jouvin-Marche, E.; Viret, C.; Faure, M.; Perron, H.; Marche, P.N. The Envelope Protein of a Human Endogenous Retrovirus-W Family Activates Innate Immunity through CD14/TLR4 and Promotes Th1-like Responses. J. Immunol. 2006, 176, 7636–7644. [Google Scholar] [CrossRef]
- Maaser, C.; Sturm, A.; Vavricka, S.R.; Kucharzik, T.; Fiorino, G.; Annese, V.; Calabrese, E.; Baumgart, D.C.; Bettenworth, D.; Borralho Nunes, P.; et al. ECCO-ESGAR Guideline for Diagnostic Assessment in IBD Part 1: Initial Diagnosis, Monitoring of Known IBD, Detection of Complications. J. Crohns Colitis 2019, 13, 144–164. [Google Scholar] [CrossRef]
- Gambarino, S.; Galliano, I.; Clemente, A.; Calvi, C.; Montanari, P.; Pau, A.; Dini, M.; Bergallo, M. Characteristics of RNA Stabilizer RNApro for Peripheral Blood Collection. Diagnostics 2024, 14, 971. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Spinsanti, G.; Zannolli, R.; Panti, C.; Ceccarelli, I.; Marsili, L.; Bachiocco, V.; Frati, F.; Aloisi, A.M. Quantitative Real-Time PCR Detection of TRPV1-4 Gene Expression in Human Leukocytes from Healthy and Hyposensitive Subjects. Mol. Pain 2008, 4, 51. [Google Scholar] [CrossRef] [PubMed]
- Biosafety and Biosecurity Policy. National Institutes of Health (NIH)—Office of Science Policy. Available online: https://osp.od.nih.gov/policies/biosafety-and-biosecurity-policy/ (accessed on 5 July 2024).
- Laboratory Biosafety Guidance Related to Coronavirus Disease (COVID-19): Interim Guidance. 28 January 2021. Available online: https://www.who.int/publications/i/item/WHO-WPE-GIH-2021.1 (accessed on 7 July 2024).
- Harvey, R.F.; Bradshaw, J.M. A Simple Index of Crohn’s-Disease Activity. Lancet 1980, 1, 514. [Google Scholar] [CrossRef]
- Schroeder, K.W.; Tremaine, W.J.; Ilstrup, D.M. Coated Oral 5-Aminosalicylic Acid Therapy for Mildly to Moderately Active Ulcerative Colitis. A Randomized Study. N. Engl. J. Med. 1987, 317, 1625–1629. [Google Scholar] [CrossRef]
- Fukuda, K.; Shinkai, Y. SETDB1-Mediated Silencing of Retroelements. Viruses 2020, 12, 596. [Google Scholar] [CrossRef]
- Chikuma, S.; Yamanaka, S.; Nakagawa, S.; Ueda, M.T.; Hayabuchi, H.; Tokifuji, Y.; Kanayama, M.; Okamura, T.; Arase, H.; Yoshimura, A. TRIM28 Expression on Dendritic Cells Prevents Excessive T Cell Priming by Silencing Endogenous Retrovirus. J. Immunol. 2021, 206, 1528–1539. [Google Scholar] [CrossRef]
- Manghera, M.; Ferguson-Parry, J.; Lin, R.; Douville, R.N. NF-κB and IRF1 Induce Endogenous Retrovirus K Expression via Interferon-Stimulated Response Elements in Its 5’ Long Terminal Repeat. J. Virol. 2016, 90, 9338–9349. [Google Scholar] [CrossRef]
- Brudek, T.; Christensen, T.; Aagaard, L.; Petersen, T.; Hansen, H.J.; Møller-Larsen, A. B Cells and Monocytes from Patients with Active Multiple Sclerosis Exhibit Increased Surface Expression of Both HERV-H Env and HERV-W Env, Accompanied by Increased Seroreactivity. Retrovirology 2009, 6, 104. [Google Scholar] [CrossRef]
- Nelson, P.N.; Roden, D.; Nevill, A.; Freimanis, G.L.; Trela, M.; Ejtehadi, H.D.; Bowman, S.; Axford, J.; Veitch, A.M.; Tugnet, N.; et al. Rheumatoid Arthritis Is Associated with IgG Antibodies to Human Endogenous Retrovirus Gag Matrix: A Potential Pathogenic Mechanism of Disease? J. Rheumatol. 2014, 41, 1952–1960. [Google Scholar] [CrossRef] [PubMed]
- Mameli, G.; Erre, G.L.; Caggiu, E.; Mura, S.; Cossu, D.; Bo, M.; Cadoni, M.L.; Piras, A.; Mundula, N.; Colombo, E.; et al. Identification of a HERV-K Env Surface Peptide Highly Recognized in Rheumatoid Arthritis (RA) Patients: A Cross-Sectional Case-Control Study. Clin. Exp. Immunol. 2017, 189, 127–131. [Google Scholar] [CrossRef]
- Perl, A.; Nagy, G.; Koncz, A.; Gergely, P.; Fernandez, D.; Doherty, E.; Telarico, T.; Bonilla, E.; Phillips, P.E. Molecular Mimicry and Immunomodulation by the HRES-1 Endogenous Retrovirus in SLE. Autoimmunity 2008, 41, 287–297. [Google Scholar] [CrossRef]
- Panova, V.; Attig, J.; Young, G.R.; Stoye, J.P.; Kassiotis, G. Antibody-Induced Internalisation of Retroviral Envelope Glycoproteins Is a Signal Initiation Event. PLoS Pathog. 2020, 16, e1008605. [Google Scholar] [CrossRef] [PubMed]
- Ko, E.-J.; Song, K.S.; Ock, M.S.; Choi, Y.H.; Kim, S.; Kim, H.-S.; Cha, H.-J. Expression Profiles of Human Endogenous Retrovirus (HERV)-K and HERV-R Env Proteins in Various Cancers. BMB Rep. 2021, 54, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Mullins, C.S.; Hühns, M.; Krohn, M.; Peters, S.; Cheynet, V.; Oriol, G.; Guillotte, M.; Ducrot, S.; Mallet, F.; Linnebacher, M. Generation, Characterization and Application of Antibodies Directed against HERV-H Gag Protein in Colorectal Samples. PLoS ONE 2016, 11, e0153349. [Google Scholar] [CrossRef] [PubMed]
- Vanuytsel, T.; Senger, S.; Fasano, A.; Shea-Donohue, T. Major Signaling Pathways in Intestinal Stem Cells. Biochim. Biophys. Acta 2013, 1830, 2410–2426. [Google Scholar] [CrossRef]
- Huang, C.; Martin, S.; Pfleger, C.; Du, J.; Buckner, J.H.; Bluestone, J.A.; Riley, J.L.; Ziegler, S.F. Cutting Edge: A Novel, Human-Specific Interacting Protein Couples FOXP3 to a Chromatin-Remodeling Complex That Contains KAP1/TRIM28. J. Immunol. 2013, 190, 4470–4473. [Google Scholar] [CrossRef]
- Chen, X.; Xiang, X.; Xia, W.; Li, X.; Wang, S.; Ye, S.; Tian, L.; Zhao, L.; Ai, F.; Shen, Z.; et al. Evolving Trends and Burden of Inflammatory Bowel Disease in Asia, 1990–2019: A Comprehensive Analysis Based on the Global Burden of Disease Study. J. Epidemiol. Glob. Health 2023, 13, 725–739. [Google Scholar] [CrossRef]
- Caviglia, G.P.; Garrone, A.; Bertolino, C.; Vanni, R.; Bretto, E.; Poshnjari, A.; Tribocco, E.; Frara, S.; Armandi, A.; Astegiano, M.; et al. Epidemiology of Inflammatory Bowel Diseases: A Population Study in a Healthcare District of North-West Italy. J. Clin. Med. 2023, 12, 641. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N.; Khalili, H.; Konijeti, G.G.; Higuchi, L.M.; de Silva, P.; Fuchs, C.S.; Willett, W.C.; Richter, J.M.; Chan, A.T. Long-Term Intake of Dietary Fat and Risk of Ulcerative Colitis and Crohn’s Disease. Gut 2014, 63, 776–784. [Google Scholar] [CrossRef]
- Piovani, D.; Danese, S.; Peyrin-Biroulet, L.; Nikolopoulos, G.K.; Lytras, T.; Bonovas, S. Environmental Risk Factors for Inflammatory Bowel Diseases: An Umbrella Review of Meta-Analyses. Gastroenterology 2019, 157, 647–659.e4. [Google Scholar] [CrossRef] [PubMed]
- Marangoni, K.; Dorneles, G.; da Silva, D.M.; Pinto, L.P.; Rossoni, C.; Fernandes, S.A. Diet as an Epigenetic Factor in Inflammatory Bowel Disease. World J. Gastroenterol. 2023, 29, 5618–5629. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Kalla, R.; Chen, J.; Zhao, J.; Zhou, X.; Adams, A.; Noble, A.; Ventham, N.T.; Wellens, J.; Ho, G.-T.; et al. Altered DNA Methylation within DNMT3A, AHRR, LTA/TNF Loci Mediates the Effect of Smoking on Inflammatory Bowel Disease. Nat. Commun. 2024, 15, 595. [Google Scholar] [CrossRef] [PubMed]
- Ryan, F.J.; Ahern, A.M.; Fitzgerald, R.S.; Laserna-Mendieta, E.J.; Power, E.M.; Clooney, A.G.; O’Donoghue, K.W.; McMurdie, P.J.; Iwai, S.; Crits-Christoph, A.; et al. Colonic Microbiota Is Associated with Inflammation and Host Epigenomic Alterations in Inflammatory Bowel Disease. Nat. Commun. 2020, 11, 1512. [Google Scholar] [CrossRef] [PubMed]
- Rogler, G.; Vavricka, S. Exposome in IBD: Recent Insights in Environmental Factors That Influence the Onset and Course of IBD. Inflamm. Bowel Dis. 2015, 21, 400–408. [Google Scholar] [CrossRef]
- Ferenc, K.; Sokal-Dembowska, A.; Helma, K.; Motyka, E.; Jarmakiewicz-Czaja, S.; Filip, R. Modulation of the Gut Microbiota by Nutrition and Its Relationship to Epigenetics. Int. J. Mol. Sci. 2024, 25, 1228. [Google Scholar] [CrossRef]
- Gabriel, U.; Steidler, A.; Trojan, L.; Michel, M.S.; Seifarth, W.; Fabarius, A. Smoking Increases Transcription of Human Endogenous Retroviruses in a Newly Established in Vitro Cell Model and in Normal Urothelium. AIDS Res. Hum. Retroviruses 2010, 26, 883–888. [Google Scholar] [CrossRef]
- Azébi, S.; Batsché, E.; Michel, F.; Kornobis, E.; Muchardt, C. Expression of Endogenous Retroviruses Reflects Increased Usage of Atypical Enhancers in T Cells. EMBO J. 2019, 38, e101107. [Google Scholar] [CrossRef]
- Pathak, R.; Feil, R. Environmental Effects on Chromatin Repression at Imprinted Genes and Endogenous Retroviruses. Curr. Opin. Chem. Biol. 2018, 45, 139–147. [Google Scholar] [CrossRef]
- Hartung, H.-P.; Derfuss, T.; Cree, B.A.; Sormani, M.P.; Selmaj, K.; Stutters, J.; Prados, F.; MacManus, D.; Schneble, H.-M.; Lambert, E.; et al. Efficacy and Safety of Temelimab in Multiple Sclerosis: Results of a Randomized Phase 2b and Extension Study. Mult. Scler. 2022, 28, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Giménez-Orenga, K.; Oltra, E. Human Endogenous Retrovirus as Therapeutic Targets in Neurologic Disease. Pharmaceuticals 2021, 14, 495. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, E.T.; Götte, M.; Tchesnokov, E.P.; Arnold, E.; Hagel, M.; Nichols, C.; Dossang, P.; Lamers, M.; Wan, P.; Steinbacher, S.; et al. Human Endogenous Retrovirus-K (HERV-K) Reverse Transcriptase (RT) Structure and Biochemistry Reveals Remarkable Similarities to HIV-1 RT and Opportunities for HERV-K-Specific Inhibition. Proc. Natl. Acad. Sci. USA 2022, 119, e2200260119. [Google Scholar] [CrossRef]
- Laderoute, M.P.; Giulivi, A.; Larocque, L.; Bellfoy, D.; Hou, Y.; Wu, H.-X.; Fowke, K.; Wu, J.; Diaz-Mitoma, F. The Replicative Activity of Human Endogenous Retrovirus K102 (HERV-K102) with HIV Viremia. AIDS 2007, 21, 2417–2424. [Google Scholar] [CrossRef]
- Tyagi, R.; Li, W.; Parades, D.; Bianchet, M.A.; Nath, A. Inhibition of Human Endogenous Retrovirus-K by Antiretroviral Drugs. Retrovirology 2017, 14, 21. [Google Scholar] [CrossRef]
- Vautrin, A.; Manchon, L.; Garcel, A.; Campos, N.; Lapasset, L.; Laaref, A.M.; Bruno, R.; Gislard, M.; Dubois, E.; Scherrer, D.; et al. Both Anti-Inflammatory and Antiviral Properties of Novel Drug Candidate ABX464 Are Mediated by Modulation of RNA Splicing. Sci. Rep. 2019, 9, 792. [Google Scholar] [CrossRef] [PubMed]
- Vermeire, S.; Solitano, V.; Peyrin-Biroulet, L.; Tilg, H.; Danese, S.; Ehrlich, H.; Scherrer, D.; Gineste, P.; d’Agay, L.; Sands, B.E. Obefazimod: A First-in-Class Drug for the Treatment of Ulcerative Colitis. J. Crohns Colitis 2023, 17, 1689–1697. [Google Scholar] [CrossRef]
- Piccinini, M.; Rinaudo, M.T.; Chiapello, N.; Ricotti, E.; Baldovino, S.; Mostert, M.; Tovo, P.-A. The Human 26S Proteasome Is a Target of Antiretroviral Agents. AIDS 2002, 16, 693–700. [Google Scholar] [CrossRef]
- Lewis, A.; Humphreys, D.T.; Pan-Castillo, B.; Berti, G.; Felice, C.; Gordon, H.; Gadhok, R.; Nijhuis, A.; Mehta, S.S.; Eleid, L.; et al. Epigenetic and Metabolic Reprogramming of Fibroblasts in Crohn’s Disease Strictures Reveals Histone Deacetylases as Therapeutic Targets. J. Crohns Colitis 2024, 18, 895–907. [Google Scholar] [CrossRef]
- Chen, R.; Tie, Y.; Lu, J.; Li, L.; Zeng, Z.; Chen, M.; Zhang, S. Tripartite Motif Family Proteins in Inflammatory Bowel Disease: Mechanisms and Potential for Interventions. Cell Prolif. 2022, 55, e13222. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Group A1 (CD) n = 48 | Group A2 (UC) n = 20 | Group B1 (HC) n = 104 | Group B2 (HC) n = 81 | |
|---|---|---|---|---|
| Median age (IQR) | 46.3 years (33.3–56.2) | 55.9 years (42.8–65.8) | 41.5 years (34.7–55.4) | 41.0 years (33.9–52.7) |
| Males n (%) | 26 (53.1) | 12 (60.0) | 62 (59.6) | 48 (59.3) |
| Duration of disease (yrs) (IQR) | 8 (3–20.3) | 8.5 (5–22.3) | ||
| Resection n (%) | 19 (39.6) | 3 (15) | ||
| Clinical disease activity * | ||||
| Remission n (%) | 28 (58.3) | 12 (60) | ||
| Mild n (%) | 13 (27.1) | 8 (40) | ||
| Moderate n (%) | 6 (12.5) | - | ||
| Severe n (%) | 1 (2.1) | - | ||
| Treatment | ||||
| Mesalazine n (%) | 33 (68.8) | 12 (60) | ||
| Topic steroids n (%) | 11 (22.9) | 3 (15) | ||
| Systemic steroids n (%) | 6 (12.5) | 3 (15) | ||
| Anti-TNF n (%) | 14 (29.2) | 5 (25) | ||
| Ustekinumab n (%) | 4 (8.3) | - | ||
| Vedolizumab n (%) | 1 (2.1) | - | ||
| Anti-Jak n (%) | 1 (2.1) | 1 (5) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tovo, P.-A.; Ribaldone, D.G.; Galliano, I.; Caviglia, G.P.; Dini, M.; Veglio, V.; Calvi, C.; Montanari, P.; Pitoni, D.; Frara, S.; et al. Enhanced Transcription of Human Endogenous Retroviruses and TRIM28 Downregulation in Patients with Inflammatory Bowel Disease. Viruses 2024, 16, 1570. https://doi.org/10.3390/v16101570
Tovo P-A, Ribaldone DG, Galliano I, Caviglia GP, Dini M, Veglio V, Calvi C, Montanari P, Pitoni D, Frara S, et al. Enhanced Transcription of Human Endogenous Retroviruses and TRIM28 Downregulation in Patients with Inflammatory Bowel Disease. Viruses. 2024; 16(10):1570. https://doi.org/10.3390/v16101570
Chicago/Turabian StyleTovo, Pier-Angelo, Davide Giuseppe Ribaldone, Ilaria Galliano, Gian Paolo Caviglia, Maddalena Dini, Valentina Veglio, Cristina Calvi, Paola Montanari, Demis Pitoni, Simone Frara, and et al. 2024. "Enhanced Transcription of Human Endogenous Retroviruses and TRIM28 Downregulation in Patients with Inflammatory Bowel Disease" Viruses 16, no. 10: 1570. https://doi.org/10.3390/v16101570
APA StyleTovo, P.-A., Ribaldone, D. G., Galliano, I., Caviglia, G. P., Dini, M., Veglio, V., Calvi, C., Montanari, P., Pitoni, D., Frara, S., Tribocco, E., Poshnjari, A., & Bergallo, M. (2024). Enhanced Transcription of Human Endogenous Retroviruses and TRIM28 Downregulation in Patients with Inflammatory Bowel Disease. Viruses, 16(10), 1570. https://doi.org/10.3390/v16101570