
Susceptibility and Resistance of SARS-CoV-2 Variants to LCB1 and Its Multivalent Derivatives


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmids and Cell Lines
2.2. Expression and Purification of Multivalent LCB1 Inhibitors
2.3. Pseudovirus Inhibition Assay
2.4. Live SARS-CoV-2 Virus Inhibition Assay
2.5. Flow Cytometry Assay
3. Results
3.1. Construction of Multivalent LCB1 Proteins with Improved Anti-SARS-CoV-2 Activity
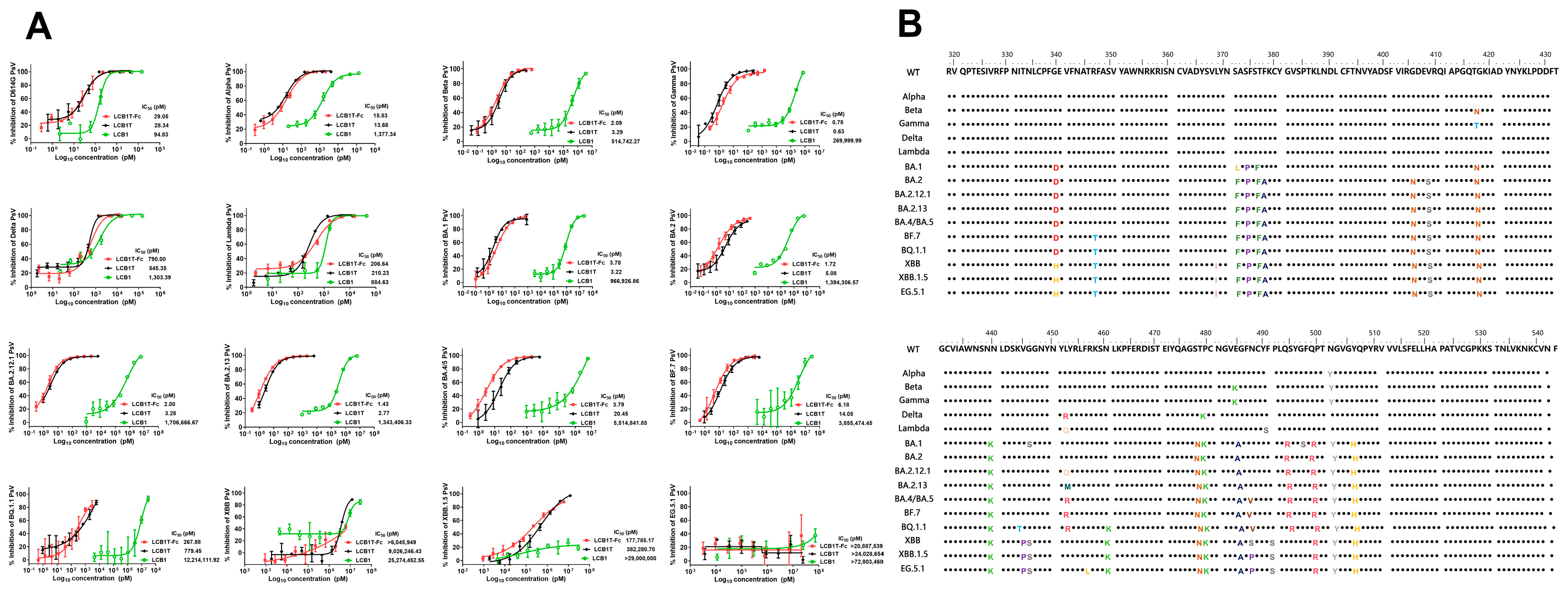
3.2. Varied Sensitivities of Diverse SARS-CoV-2 Variants to Multivalent LCB1 Inhibitor
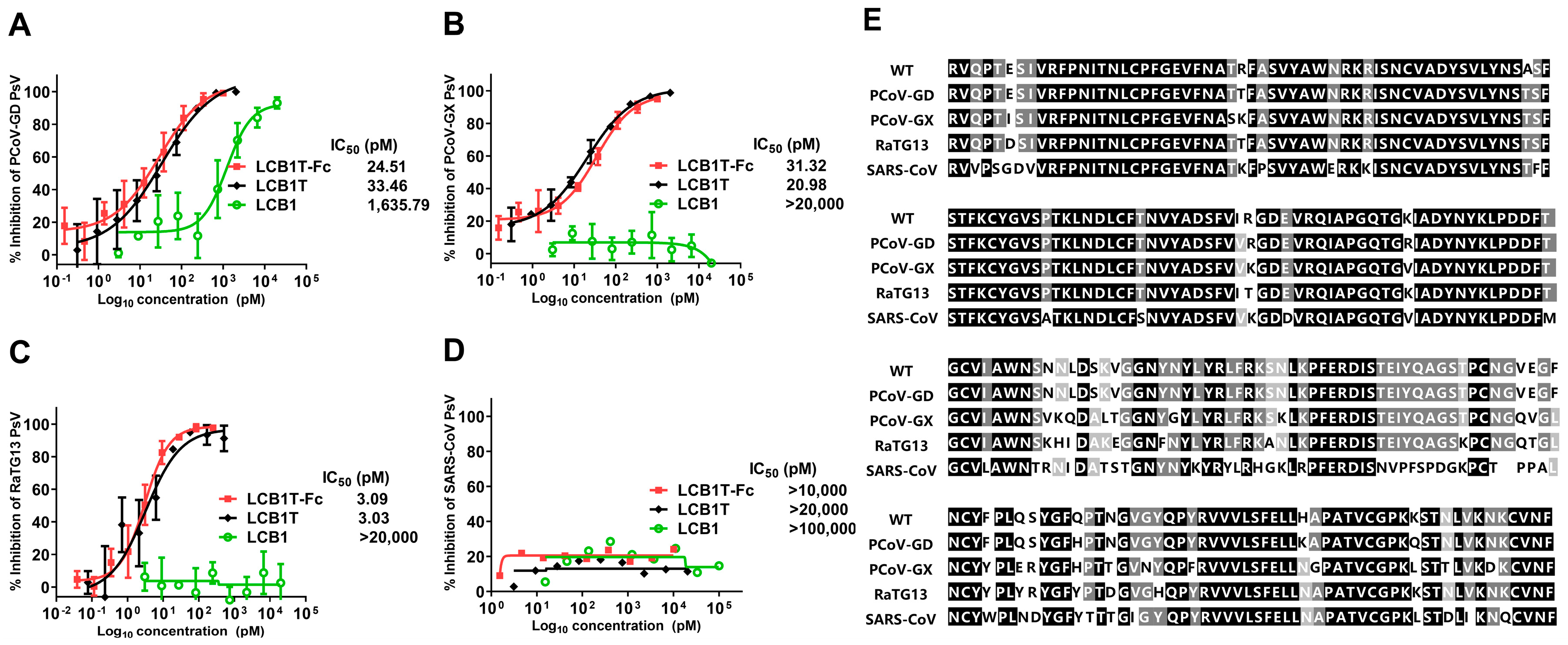
3.3. High Sensitivity of Pangolin and Bat CoVs to Multivalent LCB1 Proteins
3.4. Omicron XBB Variant Greatly Impairs the Binding Affinity of Multivalent LCB1 Protein
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Faraone, J.N.; Qu, P.; Evans, J.P.; Zheng, Y.M.; Carlin, C.; Anghelina, M.; Stevens, P.; Fernandez, S.; Jones, D.; Lozanski, G.; et al. Neutralization escape of Omicron XBB, BR.2, and BA.2.3.20 subvariants. Cell Rep. Med. 2023, 4, 101049. [Google Scholar] [CrossRef] [PubMed]
- Feikin, D.R.; Higdon, M.M.; Andrews, N.; Collie, S.; Deloria Knoll, M.; Kwong, J.C.; Link-Gelles, R.; Pilishvili, T.; Patel, M.K. Assessing COVID-19 vaccine effectiveness against Omicron subvariants: Report from a meeting of the World Health Organization. Vaccine 2023, 41, 2329–2338. [Google Scholar] [CrossRef] [PubMed]
- Tian, D.; Nie, W.; Sun, Y.; Ye, Q. The Epidemiological Features of the SARS-CoV-2 Omicron Subvariant BA.5 and Its Evasion of the Neutralizing Activity of Vaccination and Prior Infection. Vaccines 2022, 10, 1699. [Google Scholar] [CrossRef] [PubMed]
- Xiang, T.; Wang, J.; Zheng, X. The humoral and cellular immune evasion of SARS-CoV-2 Omicron and sub-lineages. Virol. Sin. 2022, 37, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Zhu, K.L.; Jiang, X.L.; Wang, X.J.; Zhan, B.D.; Gao, H.X.; Geng, X.Y.; Duan, L.J.; Dai, E.H.; Ma, M.J. Omicron subvariants escape antibodies elicited by vaccination and BA.2.2 infection. Lancet Infect. Dis. 2022, 22, 1116–1117. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020, 17, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.T.; Zhang, S.; Wang, Q.; Anang, S.; Wang, J.; Ding, H.; Kappes, J.C.; Sodroski, J. Spike glycoprotein and host cell determinants of SARS-CoV-2 entry and cytopathic effects. J. Virol. 2021, 95, e02304-20. [Google Scholar] [CrossRef]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef]
- Barton, M.I.; MacGowan, S.A.; Kutuzov, M.A.; Dushek, O.; Barton, G.J.; van der Merwe, P.A. Effects of common mutations in the SARS-CoV-2 Spike RBD and its ligand, the human ACE2 receptor on binding affinity and kinetics. Elife 2021, 10, e70658. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.S.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Goreshnik, I.; Coventry, B.; Case, J.B.; Miller, L.; Kozodoy, L.; Chen, R.E.; Carter, L.; Walls, A.C.; Park, Y.J.; et al. De novo design of picomolar SARS-CoV-2 miniprotein inhibitors. Science 2020, 370, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Case, J.B.; Chen, R.E.; Cao, L.; Ying, B.; Winkler, E.S.; Johnson, M.; Goreshnik, I.; Pham, M.N.; Shrihari, S.; Kafai, N.M.; et al. Ultrapotent miniproteins targeting the SARS-CoV-2 receptor-binding domain protect against infection and disease. Cell Host Microbe 2021, 29, 1151–1161.e1155. [Google Scholar] [CrossRef] [PubMed]
- Javanmardi, K.; Chou, C.W.; Terrace, C.I.; Annapareddy, A.; Kaoud, T.S.; Guo, Q.; Lutgens, J.; Zorkic, H.; Horton, A.P.; Gardner, E.C.; et al. Rapid characterization of spike variants via mammalian cell surface display. Mol. Cell 2021, 81, 5099–5111.e5098. [Google Scholar] [CrossRef] [PubMed]
- Hunt, A.C.; Case, J.B.; Park, Y.J.; Cao, L.; Wu, K.; Walls, A.C.; Liu, Z.; Bowen, J.E.; Yeh, H.W.; Saini, S.; et al. Multivalent designed proteins neutralize SARS-CoV-2 variants of concern and confer protection against infection in mice. Sci. Transl. Med. 2022, 14, eabn1252. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Zhu, Y.; Liu, N.; Hu, Y.; Chong, H.; He, Y. Resistance profile and mechanism of severe acute respiratory syndrome coronavirus-2 variants to LCB1 inhibitor targeting the spike receptor-binding motif. Front. Microbiol. 2022, 13, 1022006. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, M.; Liu, N.; Wu, T.; Han, X.; Zhao, G.; He, Y. Development of highly effective LCB1-based lipopeptides targeting the spike receptor-binding motif of SARS-CoV-2. Antivir. Res. 2023, 211, 105541. [Google Scholar] [CrossRef] [PubMed]
- Silverman, J.; Liu, Q.; Bakker, A.; To, W.; Duguay, A.; Alba, B.M.; Smith, R.; Rivas, A.; Li, P.; Le, H.; et al. Multivalent avimer proteins evolved by exon shuffling of a family of human receptor domains. Nat. Biotechnol. 2005, 23, 1556–1561. [Google Scholar] [CrossRef]
- Detalle, L.; Stohr, T.; Palomo, C.; Piedra, P.A.; Gilbert, B.E.; Mas, V.; Millar, A.; Power, U.F.; Stortelers, C.; Allosery, K.; et al. Generation and Characterization of ALX-0171, a Potent Novel Therapeutic Nanobody for the Treatment of Respiratory Syncytial Virus Infection. Antimicrob. Agents Chemother. 2016, 60, 6–13. [Google Scholar] [CrossRef]
- Strauch, E.M.; Bernard, S.M.; La, D.; Bohn, A.J.; Lee, P.S.; Anderson, C.E.; Nieusma, T.; Holstein, C.A.; Garcia, N.K.; Hooper, K.A.; et al. Computational design of trimeric influenza-neutralizing proteins targeting the hemagglutinin receptor binding site. Nat. Biotechnol. 2017, 35, 667–671. [Google Scholar] [CrossRef]
- Linsky, T.W.; Vergara, R.; Codina, N.; Nelson, J.W.; Walker, M.J.; Su, W.; Barnes, C.O.; Hsiang, T.Y.; Esser-Nobis, K.; Yu, K.; et al. De novo design of potent and resilient hACE2 decoys to neutralize SARS-CoV-2. Science 2020, 370, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Schoof, M.; Faust, B.; Saunders, R.A.; Sangwan, S.; Rezelj, V.; Hoppe, N.; Boone, M.; Billesbolle, C.B.; Puchades, C.; Azumaya, C.M.; et al. An ultrapotent synthetic nanobody neutralizes SARS-CoV-2 by stabilizing inactive Spike. Science 2020, 370, 1473–1479. [Google Scholar] [CrossRef] [PubMed]
- Bracken, C.J.; Lim, S.A.; Solomon, P.; Rettko, N.J.; Nguyen, D.P.; Zha, B.S.; Schaefer, K.; Byrnes, J.R.; Zhou, J.; Lui, I.; et al. Bi-paratopic and multivalent VH domains block ACE2 binding and neutralize SARS-CoV-2. Nat. Chem. Biol. 2021, 17, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Koenig, P.A.; Das, H.; Liu, H.; Kummerer, B.M.; Gohr, F.N.; Jenster, L.M.; Schiffelers, L.D.J.; Tesfamariam, Y.M.; Uchima, M.; Wuerth, J.D.; et al. Structure-guided multivalent nanobodies block SARS-CoV-2 infection and suppress mutational escape. Science 2021, 371, eabe6230. [Google Scholar] [CrossRef] [PubMed]
- Rothenberger, S.; Hurdiss, D.L.; Walser, M.; Malvezzi, F.; Mayor, J.; Ryter, S.; Moreno, H.; Liechti, N.; Bosshart, A.; Iss, C.; et al. The trispecific DARPin ensovibep inhibits diverse SARS-CoV-2 variants. Nat. Biotechnol. 2022, 40, 1845–1854. [Google Scholar] [CrossRef] [PubMed]
- Li, M.X.; Ren, Y.F.; Aw, Z.Q.; Chen, B.; Yang, Z.Q.; Lei, Y.Q.; Cheng, L.; Liang, Q.T.; Hong, J.X.; Yang, Y.L.; et al. Broadly neutralizing and protective nanobodies against SARS-CoV-2 Omicron subvariants BA.1, BA.2, and BA.4/5 and diverse sarbecoviruses. Nat. Commun. 2022, 13, 7957. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, L.; Wu, J.J.; Yu, Y.L.; Liu, S.; Li, T.; Li, Q.Q.; Ding, R.X.; Wang, H.X.; Nie, J.H.; et al. A second functional furin site in the SARS-CoV-2 spike protein. Emerg. Microbes Infec. 2022, 11, 182–194. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.M.; Dong, X.J.; Liu, N.A.; Wu, T.; Chong, H.H.; Lei, X.B.; Ren, L.L.; Wang, J.W.; He, Y.X. SARS-CoV-2 fusion-inhibitory lipopeptides maintain high potency against divergent variants of concern including Omicron. Emerg. Microbes Infec. 2022, 11, 1819–1827. [Google Scholar] [CrossRef]
- Jin, H.L.; Cheng, L.; Gong, Y.N.; Zhu, Y.M.; Chong, H.H.; Zhang, Z.; He, Y.X. Design of a bifunctional pan-sarbecovirus entry inhibitor targeting the cell receptor and viral fusion protein. J. Virol. 2023, 97, e0019223. [Google Scholar] [CrossRef]
- Zhou, B.; Cheng, L.; Song, S.; Guo, H.; Shen, S.; Wang, H.; Ge, X.; Liu, L.; Ju, B.; Zhang, Z. Identification and application of a pair of noncompeting monoclonal antibodies broadly binding to the nucleocapsid proteins of SARS-CoV-2 variants including Omicron. Virol. J. 2022, 19, 96. [Google Scholar] [CrossRef]
- Weissenborn, L.; Richel, E.; Huseman, H.; Welzer, J.; Beck, S.; Schafer, S.; Sticht, H.; Uberla, K.; Eichler, J. Smaller, Stronger, More Stable: Peptide Variants of a SARS-CoV-2 Neutralizing Miniprotein. Int. J. Mol. Sci. 2022, 23, 6309. [Google Scholar] [CrossRef] [PubMed]
- Chattaraj, R.; Kim, C.Y.; Lee, D.; Hammer, D.A. Recombinant Protein Micelles to Block Transduction by SARS-CoV-2 Pseudovirus. ACS Nano 2022, 16, 17466–17477. [Google Scholar] [CrossRef] [PubMed]
- Llewellyn, G.N.; Chen, H.Y.; Rogers, G.L.; Huang, X.; Sell, P.J.; Henley, J.E.; Cannon, P.M. Comparison of SARS-CoV-2 entry inhibitors based on ACE2 receptor or engineered Spike-binding peptides. J. Virol. 2023, 97, e0068423. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Jian, F.; Wang, J.; Yu, Y.; Song, W.; Yisimayi, A.; Wang, J.; An, R.; Chen, X.; Zhang, N.; et al. Imprinted SARS-CoV-2 humoral immunity induces convergent Omicron RBD evolution. Nature 2023, 614, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Callaway, E. Coronavirus variant XBB.1.5 rises in the United States - is it a global threat? Nature 2023, 613, 222–223. [Google Scholar] [CrossRef] [PubMed]
- Graham, F. Daily briefing: Is subvariant XBB.1.5 a global threat? Nature 2023. [Google Scholar] [CrossRef]
- Qu, P.; Evans, J.P.; Faraone, J.N.; Zheng, Y.M.; Carlin, C.; Anghelina, M.; Stevens, P.; Fernandez, S.; Jones, D.; Lozanski, G.; et al. Enhanced neutralization resistance of SARS-CoV-2 Omicron subvariants BQ.1, BQ.1.1, BA.4.6, BF.7, and BA.2.75.2. Cell Host Microbe 2023, 31, 9–17.e13. [Google Scholar] [CrossRef]
- Parums, D.V. Editorial: A Rapid Global Increase in COVID-19 is Due to the Emergence of the EG.5 (Eris) Subvariant of Omicron SARS-CoV-2. Med. Sci. Monit. 2023, 29, e942244. [Google Scholar] [CrossRef]
- Wang, Q.; Iketani, S.; Li, Z.; Liu, L.; Guo, Y.; Huang, Y.; Bowen, A.D.; Liu, M.; Wang, M.; Yu, J.; et al. Alarming antibody evasion properties of rising SARS-CoV-2 BQ and XBB subvariants. Cell 2023, 186, 279–286.e278. [Google Scholar] [CrossRef]
- Singh, M.; Bansal, V.; Feschotte, C. A Single-Cell RNA Expression Map of Human Coronavirus Entry Factors. Cell Rep. 2020, 32, 108175. [Google Scholar] [CrossRef]
- Chan, K.K.; Tan, T.J.C.; Narayanan, K.K.; Procko, E. An engineered decoy receptor for SARS-CoV-2 broadly binds protein S sequence variants. Sci. Adv. 2021, 7, eabf1738. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, M.; Mekkaoui, L.; Ilca, F.T.; Akbar, Z.; Bughda, R.; Lamb, K.; Ward, K.; Parekh, F.; Karattil, R.; Allen, C.; et al. Characterization of a Novel ACE2-Based Therapeutic with Enhanced Rather than Reduced Activity against SARS-CoV-2 Variants. J. Virol. 2021, 95, e0068521. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, Y.; Suzuki, T.; Arimori, T.; Ikemura, N.; Mihara, E.; Kirita, Y.; Ohgitani, E.; Mazda, O.; Motooka, D.; Nakamura, S.; et al. Engineered ACE2 receptor therapy overcomes mutational escape of SARS-CoV-2. Nat. Commun. 2021, 12, 3802. [Google Scholar] [CrossRef] [PubMed]
- Arimori, T.; Ikemura, N.; Okamoto, T.; Takagi, J.; Standley, D.M.; Hoshino, A. Engineering ACE2 decoy receptors to combat viral escapability. Trends Pharmacol. Sci. 2022, 43, 838–851. [Google Scholar] [CrossRef]
- Wang, B.; Zhao, J.; Liu, S.; Feng, J.; Luo, Y.; He, X.; Wang, Y.; Ge, F.; Wang, J.; Ye, B.; et al. ACE2 decoy receptor generated by high-throughput saturation mutagenesis efficiently neutralizes SARS-CoV-2 and its prevalent variants. Emerg. Microbes Infect. 2022, 11, 1488–1499. [Google Scholar] [CrossRef]
- Zhang, H.; Hu, B.; Lv, P.; Liu, Y.; Guo, M.; Wu, Z.; Zhou, K.; Dai, M.; Yu, X.; Liu, Z.; et al. An ACE2-Based Decoy Inhibitor Effectively Neutralizes SARS-CoV-2 Omicron BA.5 Variant. Viruses 2022, 14, 2387. [Google Scholar] [CrossRef]
- Liu, H.; Wu, L.; Liu, B.; Xu, K.; Lei, W.; Deng, J.; Rong, X.; Du, P.; Wang, L.; Wang, D.; et al. Two pan-SARS-CoV-2 nanobodies and their multivalent derivatives effectively prevent Omicron infections in mice. Cell Rep. Med. 2023, 4, 100918. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SARS-CoV-2 | Mean IC50 ± SD (pM) | |||||
|---|---|---|---|---|---|---|
| LCB1 | LCB1T | LCB1T-Fc | ||||
| Fold Change | Fold Change | Fold Change | ||||
| D614G | 94.83 ± 16.54 | 1 | 28.34 ± 1.33 | 1 | 29.05 ± 8.61 | 1 |
| Alpha (B.1.1.7) | 1377.34 ± 261.64 | 14.52 | 13.68 ± 0.98 | 0.48 | 15.83 ± 1.15 | 0.54 |
| Beta (B.1.351) | 514,742.27 ± 133,785.13 | 5428.05 | 3.29 ± 0.80 | 0.12 | 2.09 ± 0.14 | 0.07 |
| Gamma (B.1.1.28) | 269,999.99 ± 29,749.62 | 2847.20 | 0.63 ± 0.03 | 0.02 | 0.78 ± 0.024 | 0.03 |
| Delta (B.1.617.2) | 1303.39 ± 239.50 | 13.74 | 545.35 ± 33.10 | 19.24 | 790.00 ± 83.00 | 27.19 |
| Lambda (C.37) | 884.63 ± 222.62 | 9.33 | 210.23 ± 13.16 | 7.42 | 206.64 ± 24.76 | 7.11 |
| Omicron variants | ||||||
| BA.1 | 966,926.86 ± 43,184.25 | 10,196.42 | 3.22 ± 0.24 | 0.11 | 3.78 ± 0.63 | 0.13 |
| BA.2 | 1,394,306.57 ± 268,000.37 | 14,703.22 | 5.08 ± 0.61 | 0.18 | 1.72 ± 0.38 | 0.06 |
| BA.2.12.1 | 1,706,666.67 ± 321,601.61 | 17,997.12 | 3.28 ± 0.60 | 0.12 | 2.00 ± 0.44 | 0.07 |
| BA.2.13 | 1,343,406.33 ± 11,023.91 | 14,166.47 | 2.77 ± 0.19 | 0.10 | 1.43 ± 0.10 | 0.05 |
| BA.4/BA.5 | 5,514,841.85 ± 70,199.60 | 58,155.03 | 20.45 ± 4.91 | 0.72 | 3.79 ± 0.92 | 0.13 |
| BF.7 | 3,855,474.45 ± 532.55 | 40,656.70 | 14.05 ± 4.79 | 0.50 | 6.18 ± 2.24 | 0.21 |
| BQ.1.1 | 12,214,111.92 ± 4405.28 | 128,800.08 | 779.45 ± 65.99 | 27.50 | 267.88 ± 107.25 | 9.22 |
| XBB | 25,274,452.55 ± 2,835,889.33 | 266,523.81 | 9,026,246.43 ± 996,079.15 | 318,498.46 | >6,045,949 | >208,122.17 |
| XBB.1.5 EG.5.1 | >29,000,000 >72,803,460 | >305,810.40 >767,726.04 | 382,280.70 ± 75,662.99 >24,028,654 | 13,489.06 >847,870.64 | 177,785.17 ± 13,059.25 >20,887,539 | 6119.97 >719,020.28 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, H.; Gong, Y.; Cheng, L.; Zhu, Y.; Zhang, Z.; He, Y. Susceptibility and Resistance of SARS-CoV-2 Variants to LCB1 and Its Multivalent Derivatives. Viruses 2024, 16, 36. https://doi.org/10.3390/v16010036
Jin H, Gong Y, Cheng L, Zhu Y, Zhang Z, He Y. Susceptibility and Resistance of SARS-CoV-2 Variants to LCB1 and Its Multivalent Derivatives. Viruses. 2024; 16(1):36. https://doi.org/10.3390/v16010036
Chicago/Turabian StyleJin, Hongliang, Yani Gong, Lin Cheng, Yuanmei Zhu, Zheng Zhang, and Yuxian He. 2024. "Susceptibility and Resistance of SARS-CoV-2 Variants to LCB1 and Its Multivalent Derivatives" Viruses 16, no. 1: 36. https://doi.org/10.3390/v16010036
APA StyleJin, H., Gong, Y., Cheng, L., Zhu, Y., Zhang, Z., & He, Y. (2024). Susceptibility and Resistance of SARS-CoV-2 Variants to LCB1 and Its Multivalent Derivatives. Viruses, 16(1), 36. https://doi.org/10.3390/v16010036