

A Single Amino Acid Substitution in the Transmembrane Domain of Glycoprotein H Functionally Compensates for the Absence of gL in Pseudorabies Virus
 , , ,
, , , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Passaging Experiment
2.3. In Vitro Replication Properties
2.4. Sequencing and Site Directed Mutagenesis
2.5. Transient Transfection-Based Fusion Assay
2.6. Western Blotting
2.7. Semiquantitative Analyses of gH Surface Expression
2.8. Generation of PrV-gH I662S and PrV-gH I662S/ΔgL
2.9. Statistical Analyses
3. Results
3.1. Isolation and Characterization of PrV-ΔgLPassV99
3.2. Mutations in the gH TMD Are Reponsible for gL-Independent Fusion
3.3. The Isoleucine to Serine Substitution in the Transmembrane Domain of gH Is Sufficient to Compensate for Absence of gL
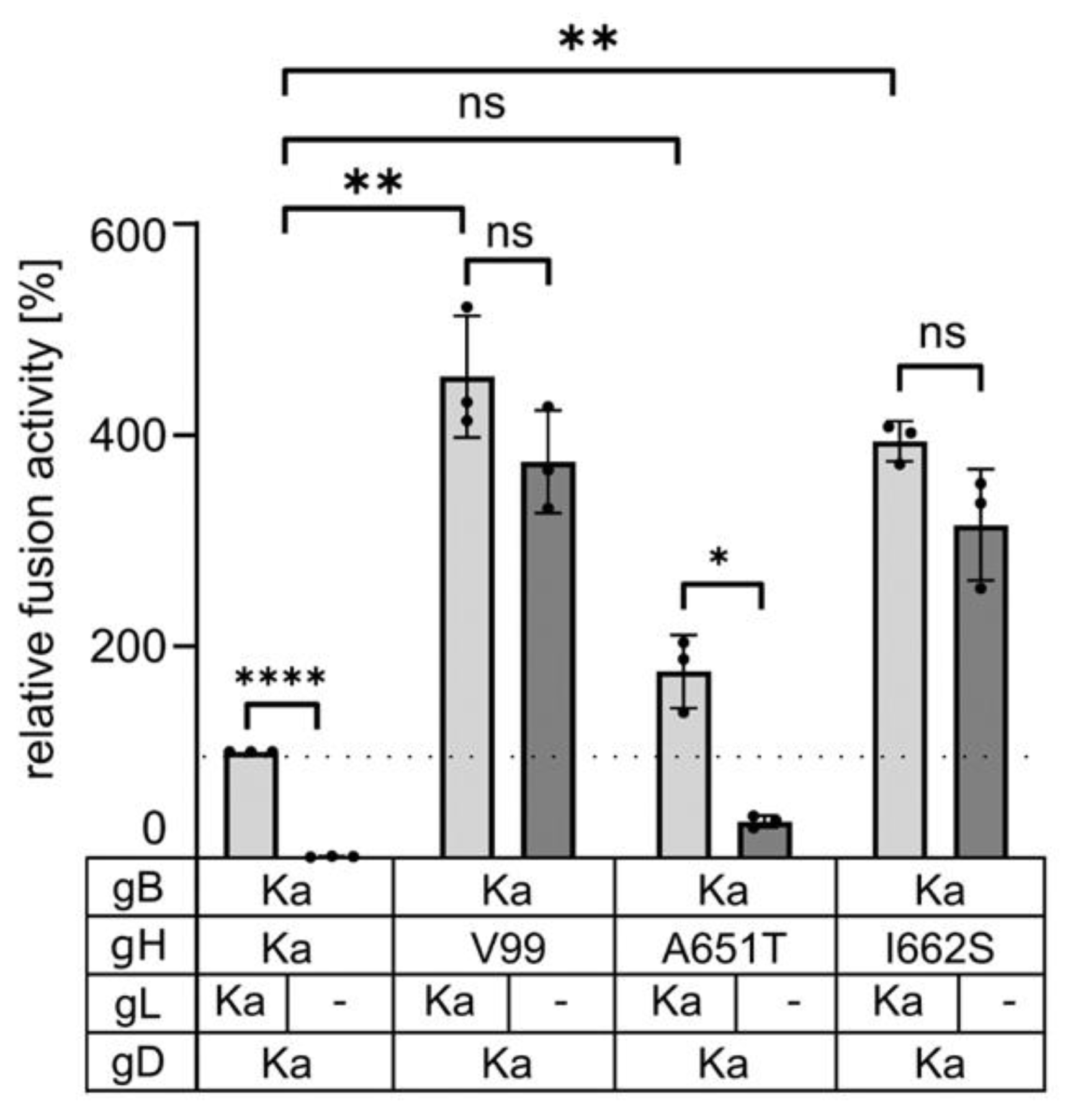
3.4. Serine, Alanine, Cysteine and Threonine at Position 662 in the gH Transmembrane Domain Can Efficiently Compensate for gL Function in Cell–cell Fusion
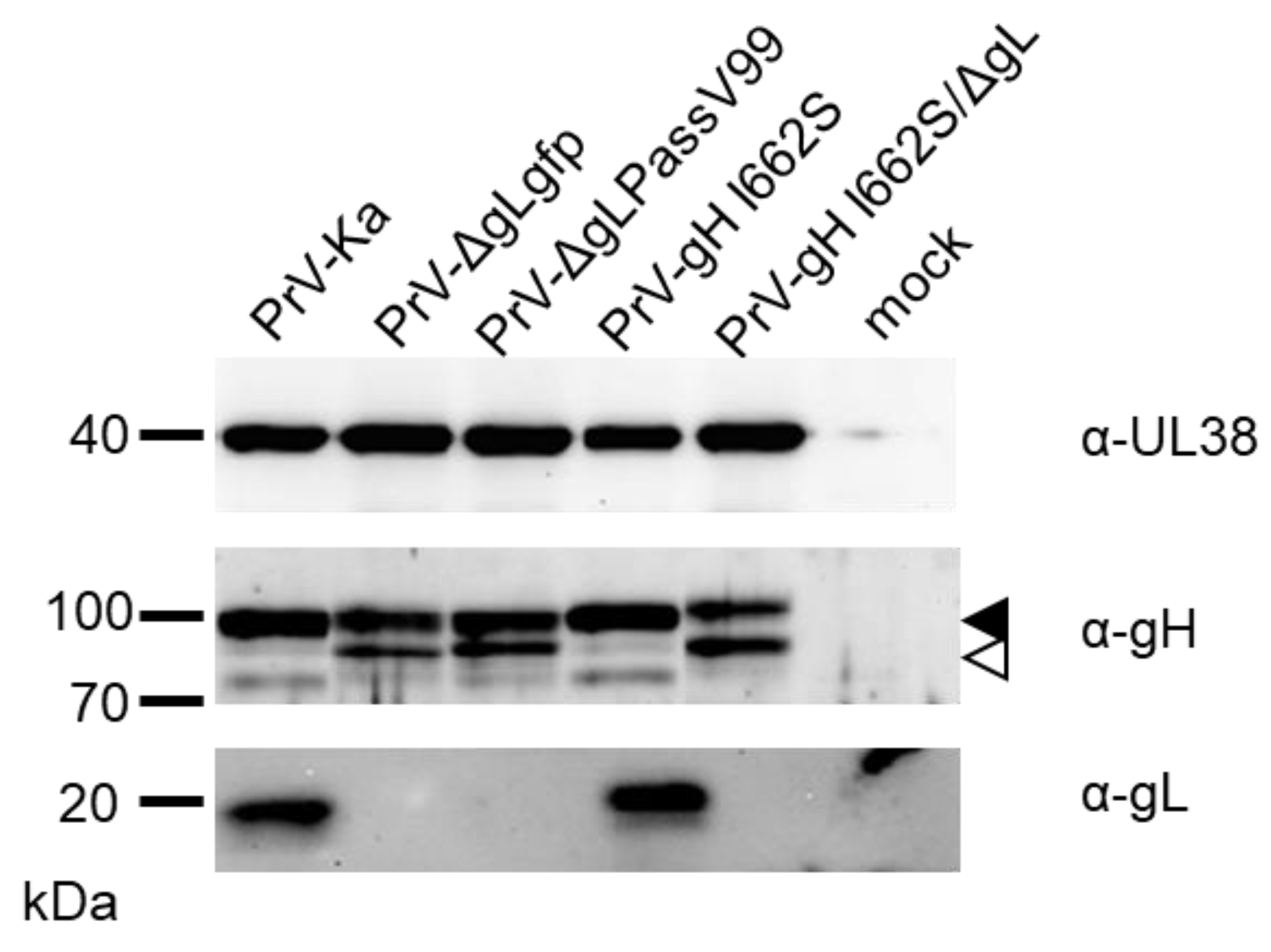
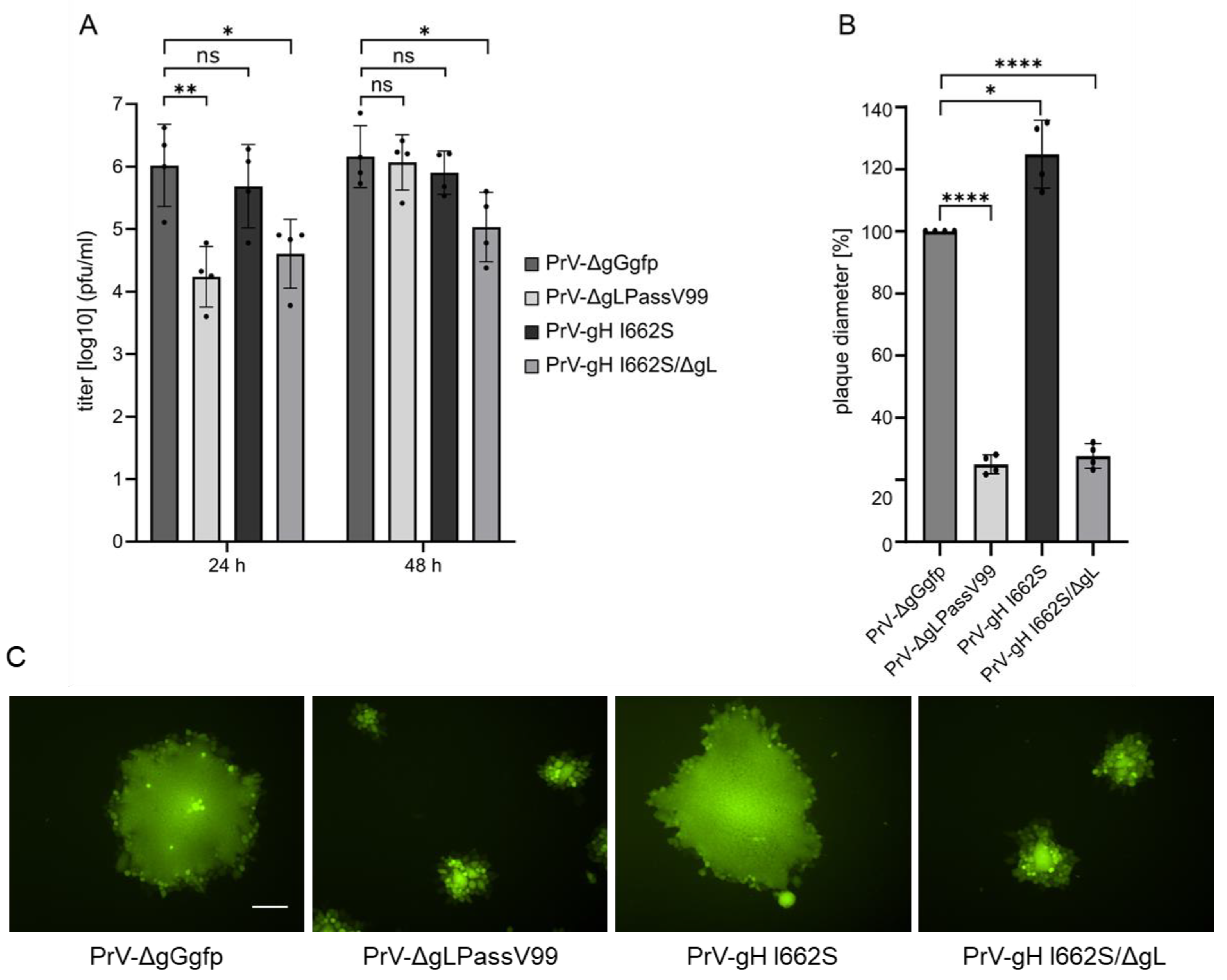
3.5. gH I662S Is Sufficient to Compensate for gL Function during Virus Entry
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Connolly, S.A.; Jardetzky, T.S.; Longnecker, R. The structural basis of herpesvirus entry. Nat. Rev. Microbiol. 2021, 19, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Cairns, T.M.; Connolly, S.A. Entry of Alphaherpesviruses. Curr. Issues Mol. Biol. 2021, 41, 63–124. [Google Scholar] [CrossRef] [PubMed]
- Vallbracht, M.; Backovic, M.; Klupp, B.G.; Rey, F.A.; Mettenleiter, T.C. Common characteristics and unique features: A comparison of the fusion machinery of the alphaherpesviruses Pseudorabies virus and Herpes simplex virus. Adv. Virus Res. 2019, 104, 225–281. [Google Scholar] [CrossRef] [PubMed]
- Gianni, T.; Massaro, R.; Campadelli-Fiume, G. Dissociation of HSV gL from gH by alphavbeta6- or alphavbeta8-integrin promotes gH activation and virus entry. Proc. Natl. Acad. Sci. USA 2015, 112, E3901–E3910. [Google Scholar] [CrossRef] [PubMed]
- Backovic, M.; DuBois, R.M.; Cockburn, J.J.; Sharff, A.J.; Vaney, M.C.; Granzow, H.; Klupp, B.G.; Bricogne, G.; Mettenleiter, T.C.; Rey, F.A. Structure of a core fragment of glycoprotein H from pseudorabies virus in complex with antibody. Proc. Natl. Acad. Sci. USA 2010, 107, 22635–22640. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, L.; Browne, H.; Wargent, V.; Davis-Poynter, N.; Primorac, S.; Goldsmith, K.; Minson, A.C.; Johnson, D.C. A novel herpes simplex virus glycoprotein, gL, forms a complex with glycoprotein H (gH) and affects normal folding and surface expression of gH. J. Virol. 1992, 66, 2240–2250. [Google Scholar] [CrossRef]
- Klupp, B.G.; Fuchs, W.; Weiland, E.; Mettenleiter, T.C. Pseudorabies virus glycoprotein L is necessary for virus infectivity but dispensable for virion localization of glycoprotein H. J. Virol. 1997, 71, 7687–7695. [Google Scholar] [CrossRef]
- Schroter, C.; Vallbracht, M.; Altenschmidt, J.; Kargoll, S.; Fuchs, W.; Klupp, B.G.; Mettenleiter, T.C. Mutations in Pseudorabies Virus Glycoproteins gB, gD, and gH Functionally Compensate for the Absence of gL. J. Virol. 2015, 90, 2264–2272. [Google Scholar] [CrossRef]
- Klupp, B.G.; Mettenleiter, T.C. Glycoprotein gL-independent infectivity of pseudorabies virus is mediated by a gD-gH fusion protein. J. Virol. 1999, 73, 3014–3022. [Google Scholar] [CrossRef]
- Klupp, B.G.; Nixdorf, R.; Mettenleiter, T.C. Pseudorabies virus glycoprotein M inhibits membrane fusion. J. Virol. 2000, 74, 6760–6768. [Google Scholar] [CrossRef]
- Cairns, T.M.; Milne, R.S.; Ponce-de-Leon, M.; Tobin, D.K.; Cohen, G.H.; Eisenberg, R.J. Structure-function analysis of herpes simplex virus type 1 gD and gH-gL: Clues from gDgH chimeras. J. Virol. 2003, 77, 6731–6742. [Google Scholar] [CrossRef] [PubMed]
- Cairns, T.M.; Friedman, L.S.; Lou, H.; Whitbeck, J.C.; Shaner, M.S.; Cohen, G.H.; Eisenberg, R.J. N-terminal mutants of herpes simplex virus type 2 gH are transported without gL but require gL for function. J. Virol. 2007, 81, 5102–5111. [Google Scholar] [CrossRef] [PubMed]
- Atanasiu, D.; Cairns, T.M.; Whitbeck, J.C.; Saw, W.T.; Rao, S.; Eisenberg, R.J.; Cohen, G.H. Regulation of herpes simplex virus gB-induced cell-cell fusion by mutant forms of gH/gL in the absence of gD and cellular receptors. mBio 2013, 4, e00046-13. [Google Scholar] [CrossRef] [PubMed]
- Atanasiu, D.; Saw, W.T.; Cohen, G.H.; Eisenberg, R.J. Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J. Virol. 2010, 84, 12292–12299. [Google Scholar] [CrossRef] [PubMed]
- Wille, P.T.; Wisner, T.W.; Ryckman, B.; Johnson, D.C. Human cytomegalovirus (HCMV) glycoprotein gB promotes virus entry in trans acting as the viral fusion protein rather than as a receptor-binding protein. mBio 2013, 4, e00332-13. [Google Scholar] [CrossRef] [PubMed]
- Pataki, Z.; Rebolledo Viveros, A.; Heldwein, E.E. Herpes Simplex Virus 1 Entry Glycoproteins Form Complexes before and during Membrane Fusion. mBio 2022, 13, e02039-22. [Google Scholar] [CrossRef]
- Silverman, J.L.; Heldwein, E.E. Mutations in the cytoplasmic tail of herpes simplex virus 1 gH reduce the fusogenicity of gB in transfected cells. J. Virol. 2013, 87, 10139–10147. [Google Scholar] [CrossRef]
- Chen, J.; Jardetzky, T.S.; Longnecker, R. The Cytoplasmic Tail Domain of Epstein-Barr Virus gH Regulates Membrane Fusion Activity through Altering gH Binding to gp42 and Epithelial Cell Attachment. mBio 2016, 7, e01871-16. [Google Scholar] [CrossRef]
- Yang, E.; Arvin, A.M.; Oliver, S.L. The cytoplasmic domain of varicella-zoster virus glycoprotein H regulates syncytia formation and skin pathogenesis. PLoS Pathog. 2014, 10, e1004173. [Google Scholar] [CrossRef]
- Vallbracht, M.; Fuchs, W.; Klupp, B.G.; Mettenleiter, T.C. Functional Relevance of the Transmembrane Domain and Cytoplasmic Tail of the Pseudorabies Virus Glycoprotein H for Membrane Fusion. J. Virol. 2018, 92, e00376-18. [Google Scholar] [CrossRef]
- Harman, A.; Browne, H.; Minson, T. The transmembrane domain and cytoplasmic tail of herpes simplex virus type 1 glycoprotein H play a role in membrane fusion. J. Virol. 2002, 76, 10708–10716. [Google Scholar] [CrossRef] [PubMed]
- Pataki, Z.; Sanders, E.K.; Heldwein, E.E. A surface pocket in the cytoplasmic domain of the herpes simplex virus fusogen gB controls membrane fusion. PLoS Pathog. 2022, 18, e1010435. [Google Scholar] [CrossRef] [PubMed]
- Rogalin, H.B.; Heldwein, E.E. Interplay between the Herpes Simplex Virus 1 gB Cytodomain and the gH Cytotail during Cell-Cell Fusion. J. Virol. 2015, 89, 12262–12272. [Google Scholar] [CrossRef] [PubMed]
- Vallbracht, M.; Lotzsch, H.; Klupp, B.G.; Fuchs, W.; Vollmer, B.; Grunewald, K.; Backovic, M.; Rey, F.A.; Mettenleiter, T.C. In Vitro Viral Evolution Identifies a Critical Residue in the Alphaherpesvirus Fusion Glycoprotein B Ectodomain That Controls gH/gL-Independent Entry. mBio 2021, 12, e00557-21. [Google Scholar] [CrossRef] [PubMed]
- Klupp, B.; Altenschmidt, J.; Granzow, H.; Fuchs, W.; Mettenleiter, T.C. Glycoproteins required for entry are not necessary for egress of pseudorabies virus. J. Virol. 2008, 82, 6299–6309. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, A.S.; Vatter, A.E. A comparison of herpes simplex and pseudorabies viruses. Virology 1959, 7, 394–407. [Google Scholar] [CrossRef]
- Rothermel, M.; Schobel, N.; Damann, N.; Klupp, B.G.; Mettenleiter, T.C.; Hatt, H.; Wetzel, C.H. Anterograde transsynaptic tracing in the murine somatosensory system using Pseudorabies virus (PrV): A “live-cell”-tracing tool for analysis of identified neurons in vitro. J. Neurovirol 2007, 13, 579–585. [Google Scholar] [CrossRef]
- Mettenleiter, T.C. Glycoprotein gIII deletion mutants of pseudorabies virus are impaired in virus entry. Virology 1989, 171, 623–625. [Google Scholar] [CrossRef]
- Bohm, S.W.; Eckroth, E.; Backovic, M.; Klupp, B.G.; Rey, F.A.; Mettenleiter, T.C.; Fuchs, W. Structure-based functional analyses of domains II and III of pseudorabies virus glycoprotein H. J. Virol. 2015, 89, 1364–1376. [Google Scholar] [CrossRef]
- Vallbracht, M.; Schroter, C.; Klupp, B.G.; Mettenleiter, T.C. Transient Transfection-based Fusion Assay for Viral Proteins. Bio-Protocol 2017, 7, e2162. [Google Scholar] [CrossRef]
- Bohm, S.W.; Backovic, M.; Klupp, B.G.; Rey, F.A.; Mettenleiter, T.C.; Fuchs, W. Functional Characterization of Glycoprotein H Chimeras Composed of Conserved Domains of the Pseudorabies Virus and Herpes Simplex Virus 1 Homologs. J. Virol. 2016, 90, 421–432. [Google Scholar] [CrossRef]
- Vallbracht, M.; Rehwaldt, S.; Klupp, B.G.; Mettenleiter, T.C.; Fuchs, W. Functional Relevance of the N-Terminal Domain of Pseudorabies Virus Envelope Glycoprotein H and Its Interaction with Glycoprotein L. J. Virol. 2017, 91, e00061-17. [Google Scholar] [CrossRef]
- Szpara, M.L.; Tafuri, Y.R.; Parsons, L.; Shamim, S.R.; Verstrepen, K.J.; Legendre, M.; Enquist, L.W. A wide extent of inter-strain diversity in virulent and vaccine strains of alphaherpesviruses. PLoS Pathog. 2011, 7, e1002282. [Google Scholar] [CrossRef] [PubMed]
- Grimm, K.S.; Klupp, B.G.; Granzow, H.; Muller, F.M.; Fuchs, W.; Mettenleiter, T.C. Analysis of viral and cellular factors influencing herpesvirus-induced nuclear envelope breakdown. J. Virol. 2012, 86, 6512–6521. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.; Gerdts, V.; Beyer, J.; Klupp, B.G.; Mettenleiter, T.C. Glycoprotein D-independent infectivity of pseudorabies virus results in an alteration of in vivo host range and correlates with mutations in glycoproteins B and H. J. Virol. 2001, 75, 10054–10064. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nixdorf, R.; Klupp, B.G.; Mettenleiter, T.C. Restoration of function of carboxy-terminally truncated pseudorabies virus glycoprotein B by point mutations in the ectodomain. J. Virol. 2001, 75, 11526–11533. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Atanasiu, D.; Saw, W.T.; Cairns, T.M.; Eisenberg, R.J.; Cohen, G.H. Using Split Luciferase Assay and anti-HSV Glycoprotein Monoclonal Antibodies to Predict a Functional Binding Site Between gD and gH/gL. J. Virol. 2021, 95, e00053-21. [Google Scholar] [CrossRef]
- Avitabile, E.; Forghieri, C.; Campadelli-Fiume, G. Cross talk among the glycoproteins involved in herpes simplex virus entry and fusion: The interaction between gB and gH/gL does not necessarily require gD. J. Virol. 2009, 83, 10752–10760. [Google Scholar] [CrossRef]
- Atanasiu, D.; Whitbeck, J.C.; Cairns, T.M.; Reilly, B.; Cohen, G.H.; Eisenberg, R.J. Bimolecular complementation reveals that glycoproteins gB and gH/gL of herpes simplex virus interact with each other during cell fusion. Proc. Natl. Acad. Sci. USA 2007, 104, 18718–18723. [Google Scholar] [CrossRef] [PubMed]
- Langosch, D.; Arkin, I.T. Interaction and conformational dynamics of membrane-spanning protein helices. Protein Sci. 2009, 18, 1343–1358. [Google Scholar] [CrossRef]
- Handler, C.G.; Eisenberg, R.J.; Cohen, G.H. Oligomeric structure of glycoproteins in herpes simplex virus type 1. J. Virol. 1996, 70, 6067–6070. [Google Scholar] [CrossRef] [PubMed]
- Bocharov, E.V.; Volynsky, P.E.; Pavlov, K.V.; Efremov, R.G.; Arseniev, A.S. Structure elucidation of dimeric transmembrane domains of bitopic proteins. Cell Adhes. Migr. 2010, 4, 284–298. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Designation | Primer Sequence (5′ -> 3′) 1 |
|---|---|
| gH A651T | C GTG GTG ACC TCC GTC GTG G |
| gH I662A | CG GCC ATC ACC GTG GGG GCC CTG TAC GCC CTA TTC |
| gH I662G | CG GCC ATC ACC GTG GGG GGC CTG TAC GCC CTA TTC |
| gH I662R | CG GCC ATC ACC GTG GGG CGC CTG TAC GCC CTA TTC |
| gH I662S | CG GCC ATC ACC GTG GGG AGC CTG TAC GCC CTA TTC |
| gH I662T | CG GCC ATC ACC GTG GGG ACC CTG TAC GCC CTA TTC |
| gH I662Y | CG GCC ATC ACC GTG GGG TAC CTG TAC GCC CTA TTC |
| gH I662C | CG GCC ATC ACC GTG GGG TGC CTG TAC GCC CTA TTC |
| gH I662V | CG GCC ATC ACC GTG GGG GTC CTG TAC GCC CTA TTC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vallbracht, M.; Schnell, M.; Seyfarth, A.; Fuchs, W.; Küchler, R.; Mettenleiter, T.C.; Klupp, B.G. A Single Amino Acid Substitution in the Transmembrane Domain of Glycoprotein H Functionally Compensates for the Absence of gL in Pseudorabies Virus. Viruses 2024, 16, 26. https://doi.org/10.3390/v16010026
Vallbracht M, Schnell M, Seyfarth A, Fuchs W, Küchler R, Mettenleiter TC, Klupp BG. A Single Amino Acid Substitution in the Transmembrane Domain of Glycoprotein H Functionally Compensates for the Absence of gL in Pseudorabies Virus. Viruses. 2024; 16(1):26. https://doi.org/10.3390/v16010026
Chicago/Turabian StyleVallbracht, Melina, Marina Schnell, Annemarie Seyfarth, Walter Fuchs, Richard Küchler, Thomas C. Mettenleiter, and Barbara G. Klupp. 2024. "A Single Amino Acid Substitution in the Transmembrane Domain of Glycoprotein H Functionally Compensates for the Absence of gL in Pseudorabies Virus" Viruses 16, no. 1: 26. https://doi.org/10.3390/v16010026
APA StyleVallbracht, M., Schnell, M., Seyfarth, A., Fuchs, W., Küchler, R., Mettenleiter, T. C., & Klupp, B. G. (2024). A Single Amino Acid Substitution in the Transmembrane Domain of Glycoprotein H Functionally Compensates for the Absence of gL in Pseudorabies Virus. Viruses, 16(1), 26. https://doi.org/10.3390/v16010026