
Genomic Analyses Uncover Evolutionary Features of Influenza A/H3N2 Viruses in Yunnan Province, China, from 2017 to 2022

Abstract
1. Introduction
2. Materials and Methods
2.1. Specimen Collection and Isolation/Identification of Viruses
2.2. Whole-Genome Sequencing of Influenza A/H3N2 Viruses
2.3. Phylogenetic Analyses of Influenza A/H3N2 Viruses
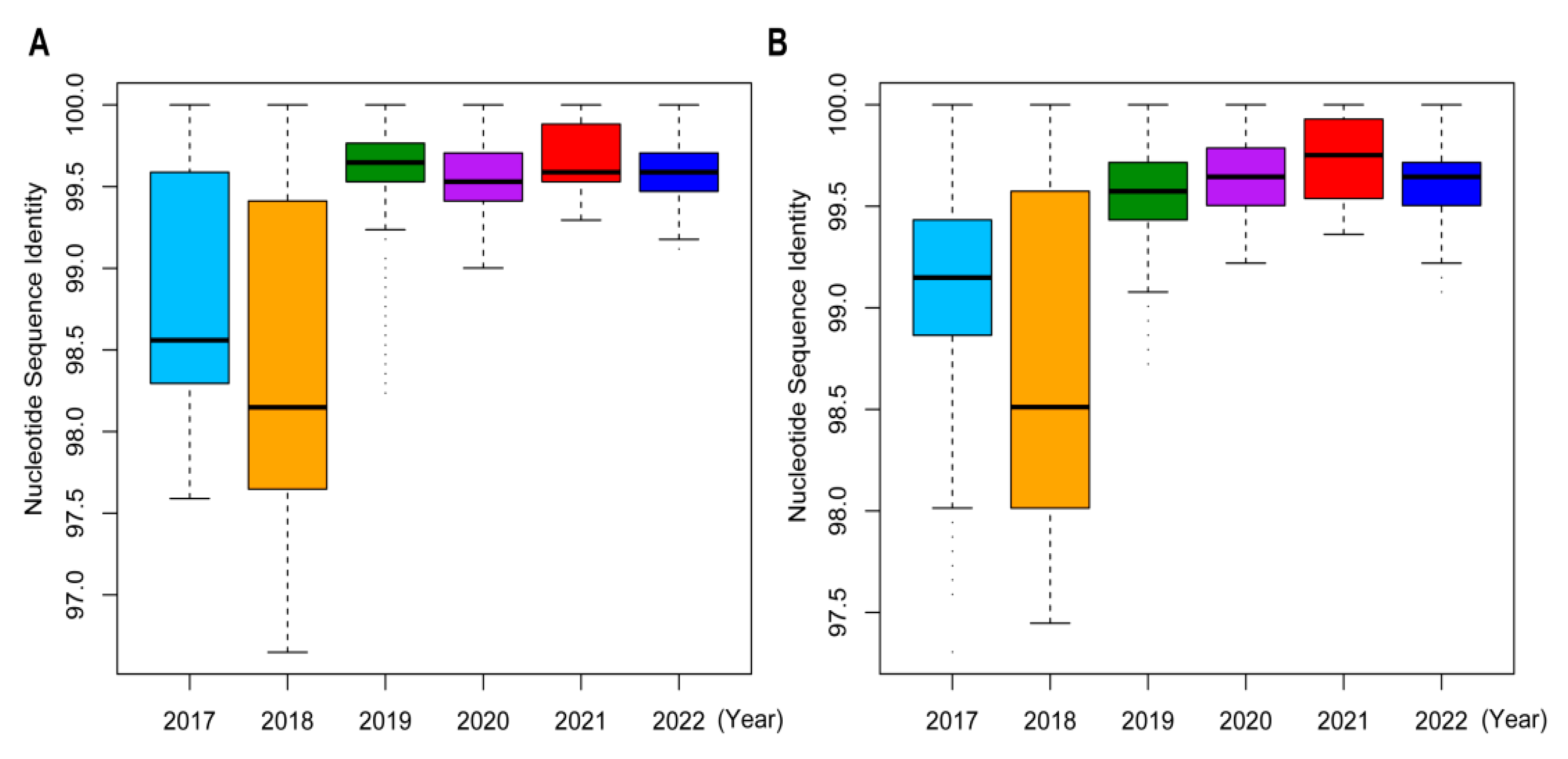
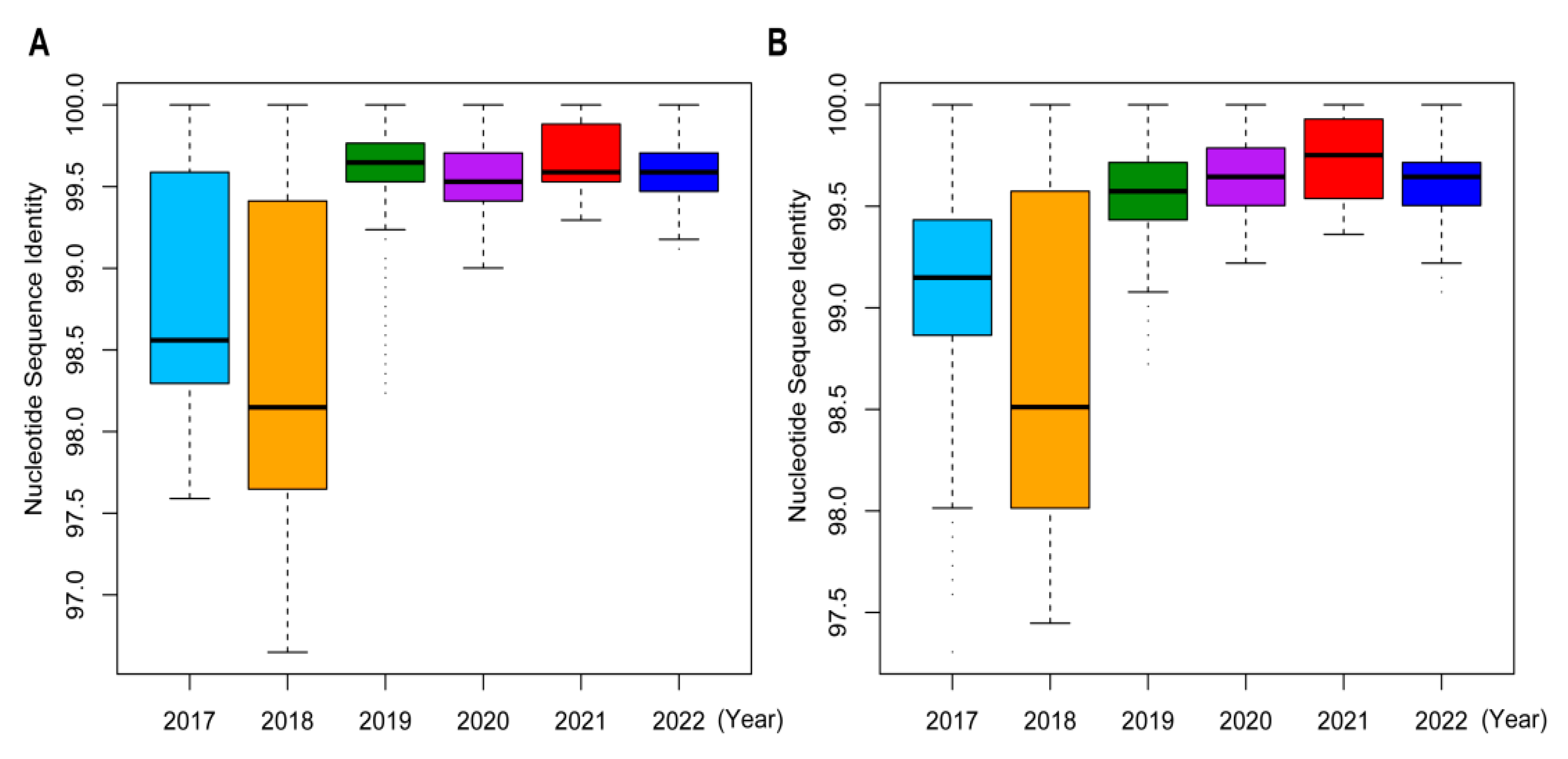
2.4. Sequence Homologies and Mutation Analyses of Influenza A/H3N2 Viruses
2.5. Selective Constraint Analyses of Influenza A/H3N2 Viruses
2.6. Vaccine Effectiveness Evaluation
3. Results
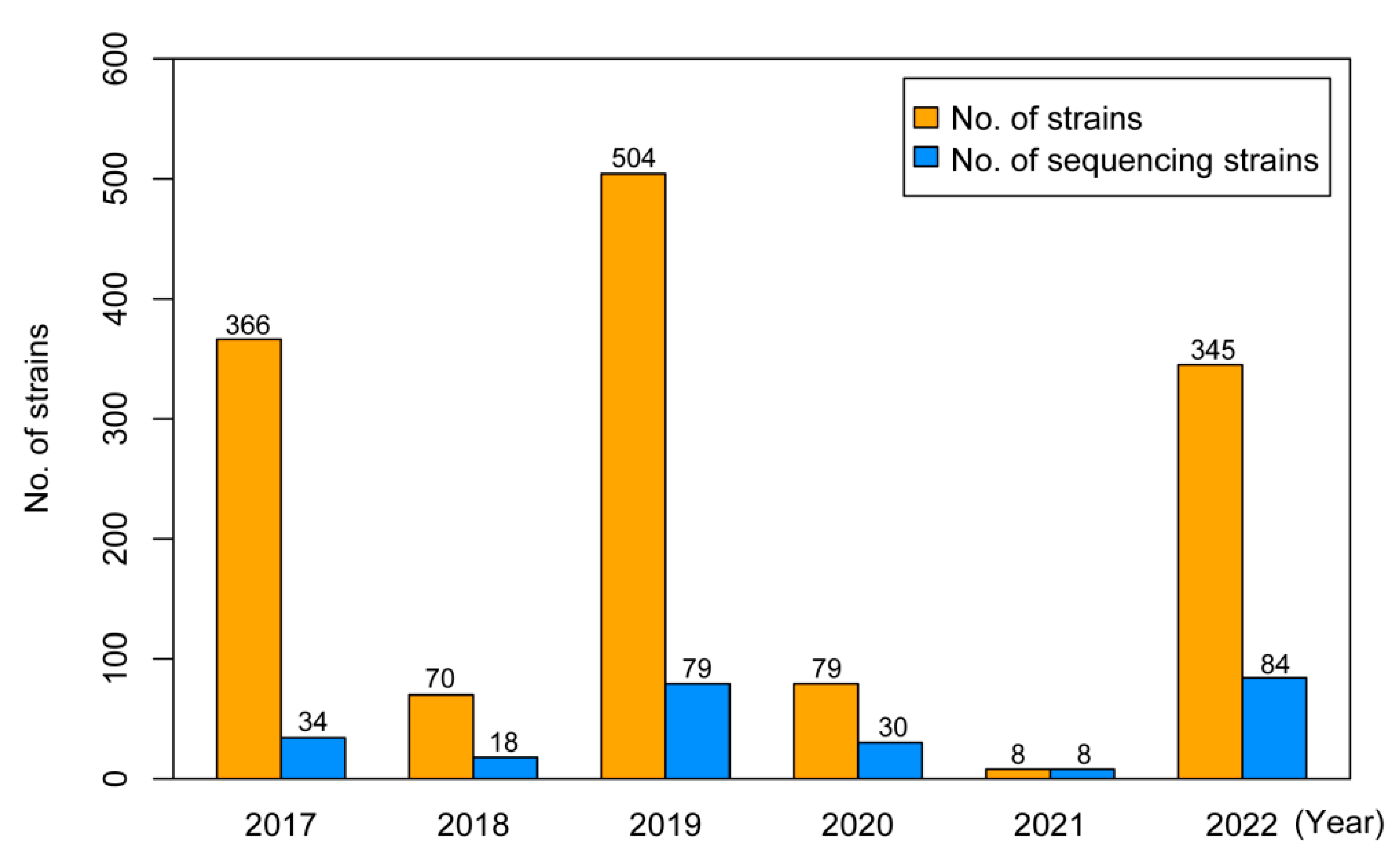
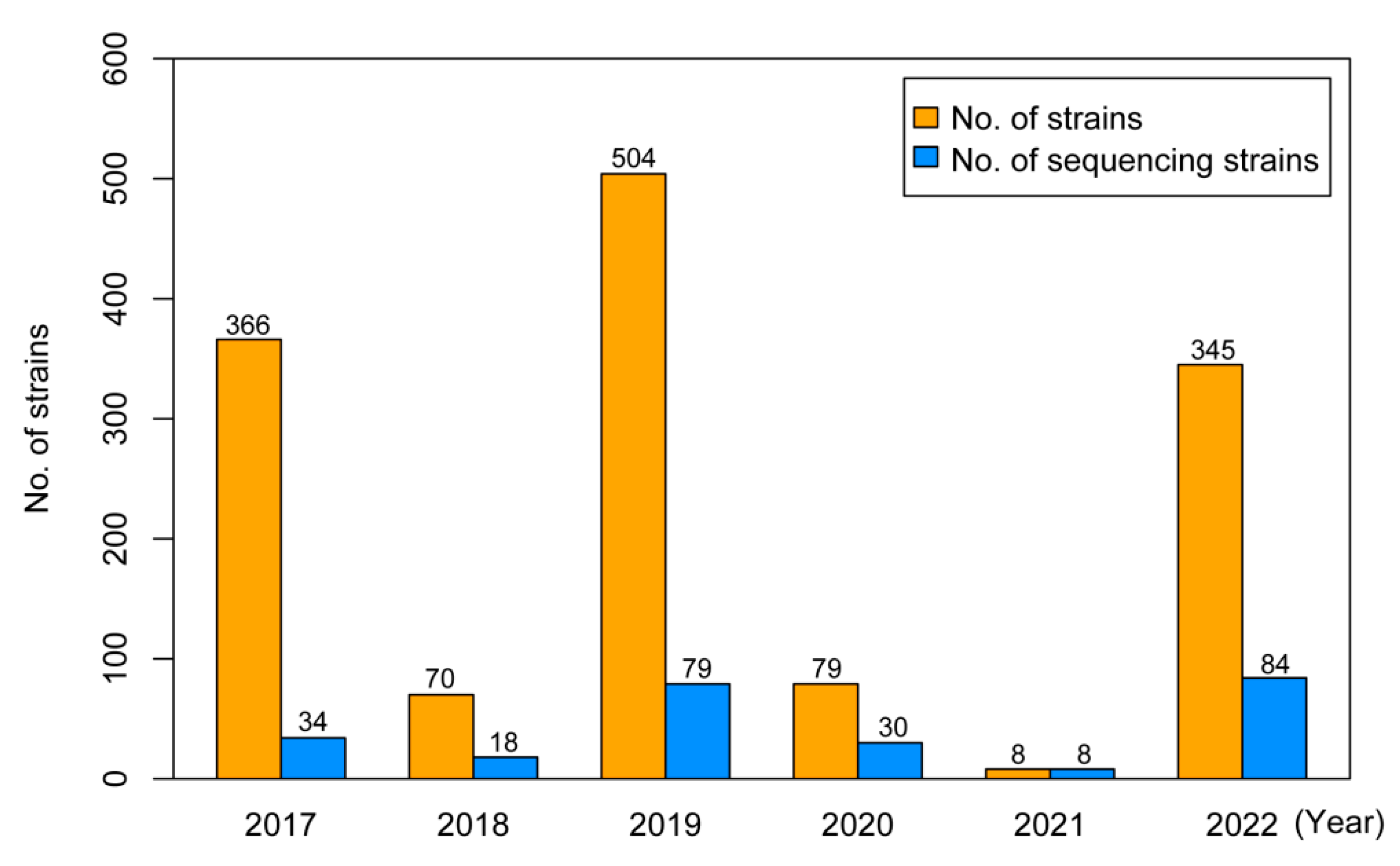
3.1. Isolation Rate and Demographic Characteristics of Influenza
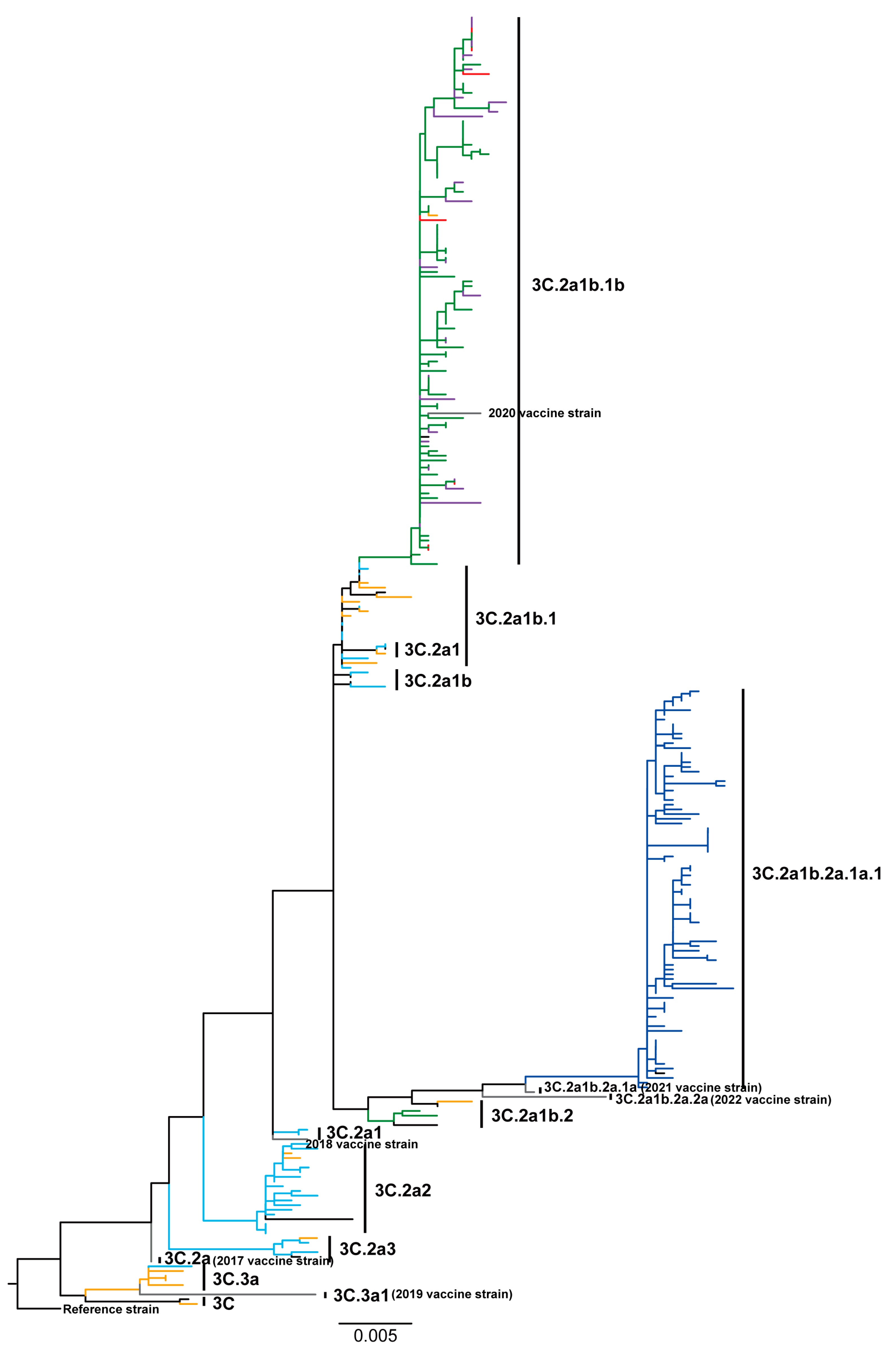
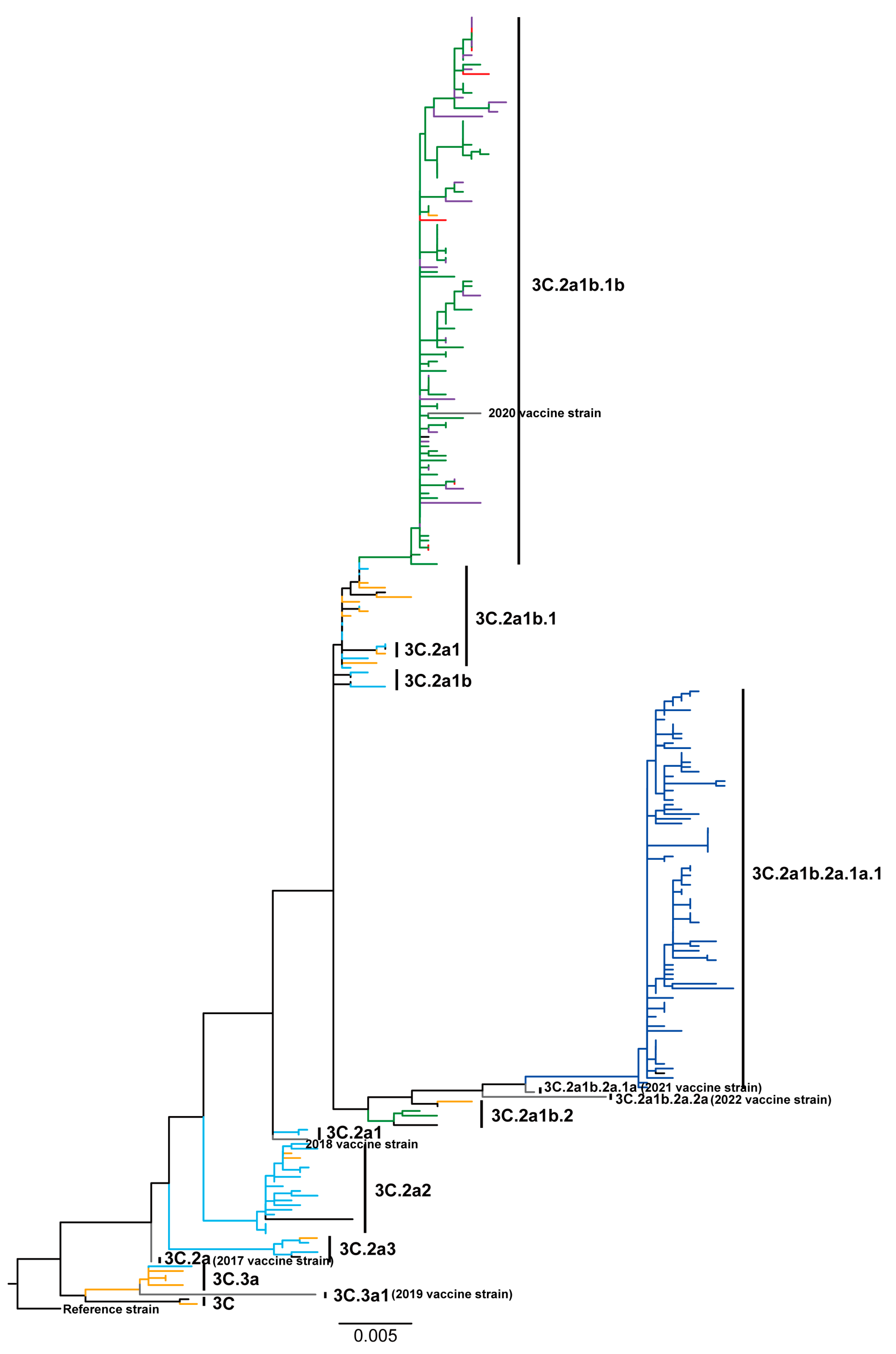
3.2. Phylogenetic Patterns Highlight High Sequence Diversity of Influenza A/H3N2 Viruses in 2018
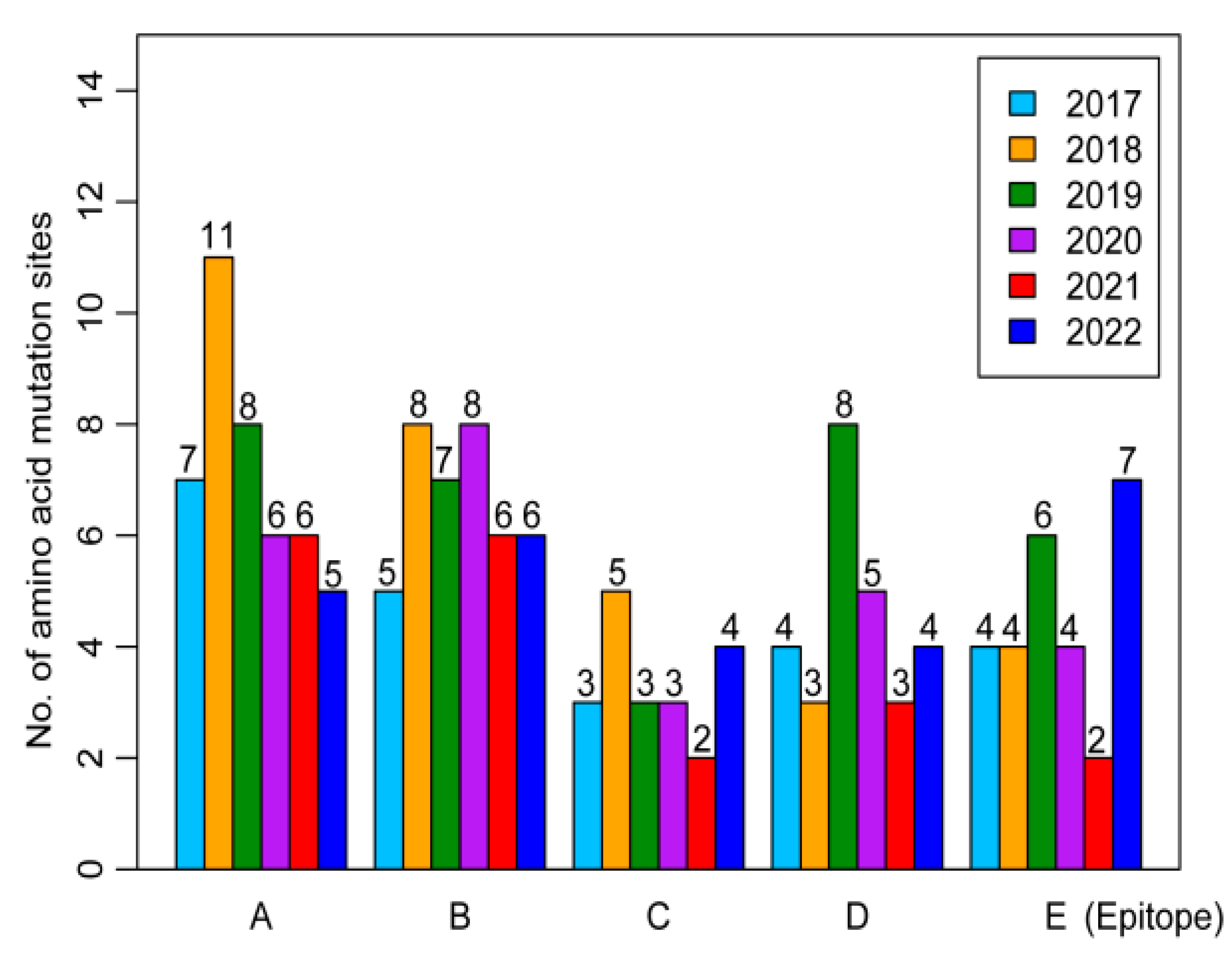
3.3. Amino Acid Mutation Analyses of Influenza A/H3N2 Viruses
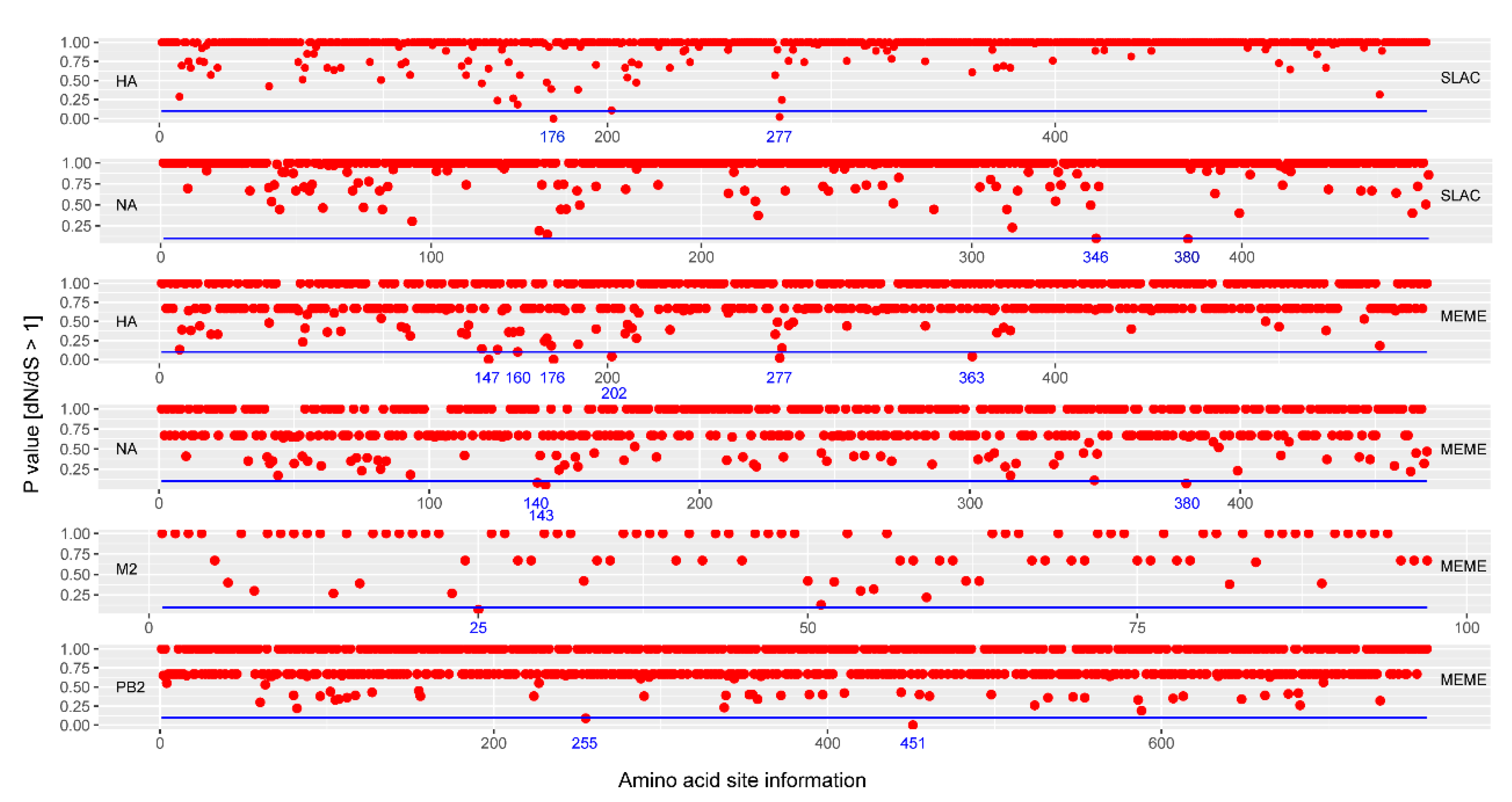
3.4. Selection Pressure Analyses of Influenza A/H3N2 Viruses
3.5. Estimation of Vaccine Efficacy for Influenza A/H3N2 Viruses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Influenza (Seasonal) Key Facts. 2023. Available online: https://www.who.int/en/news-room/fact-sheets/detail/influenza-(seasonal) (accessed on 3 October 2023).
- Feng, L.; Shay, D.K.; Jiang, Y.; Zhou, H.; Chen, X.; Zheng, Y.; Jiang, L.; Zhang, Q.; Lin, H.; Wang, S.; et al. Influenza-associated mortality in temperate and subtropical Chinese cities, 2003–2008. Bull. World Health Organ. 2012, 90, 279–288b. [Google Scholar] [CrossRef]
- Prachanronarong, K.L.; Canale, A.S.; Liu, P.; Somasundaran, M.; Hou, S.; Poh, Y.P.; Han, T.; Zhu, Q.; Renzette, N.; Zeldovich, K.B.; et al. Mutations in Influenza A Virus Neuraminidase and Hemagglutinin Confer Resistance against a Broadly Neutralizing Hemagglutinin Stem Antibody. J. Virol. 2019, 93, e01639-18. [Google Scholar] [CrossRef]
- Kim, K.; Kim, Y. Population genetic processes affecting the mode of selective sweeps and effective population size in influenza virus H3N2. BMC Evol. Biol. 2016, 16, 156. [Google Scholar] [CrossRef] [PubMed]
- Glatman-Freedman, A.; Drori, Y.; Beni, S.A.; Friedman, N.; Pando, R.; Sefty, H.; Tal, I.; McCauley, J.; Rahav, G.; Keller, N.; et al. Genetic divergence of Influenza A(H3N2) amino acid substitutions mark the beginning of the 2016–2017 winter season in Israel. J. Clin. Virol. 2017, 93, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Tang, J.W.; Loh, T.P.; Oon, L.L.; Koay, E.S. Predicting clinical severity based on substitutions near epitope A of influenza A/H3N2. Infect. Genet. Evol. 2015, 34, 292–297. [Google Scholar] [PubMed]
- Webster, R.G.; Bean, W.J.; Gorman, O.T.; Chambers, T.M.; Kawaoka, Y. Evolution and ecology of influenza A viruses. Microbiol. Rev. 1992, 56, 152–179. [Google Scholar] [CrossRef]
- Matrosovich, M.N.; Matrosovich, T.Y.; Gray, T.; Roberts, N.A.; Klenk, H.D. Neuraminidase is important for the initiation of influenza virus infection in human airway epithelium. J. Virol. 2004, 78, 12665–12667. [Google Scholar] [CrossRef]
- Wiley, D.C.; Skehel, J.J. The structure and function of the hemagglutinin membrane glycoprotein of influenza virus. Annu. Rev. Biochem. 1987, 56, 365–394. [Google Scholar] [CrossRef]
- Shih, A.C.; Hsiao, T.C.; Ho, M.S.; Li, W.H. Simultaneous amino acid substitutions at antigenic sites drive influenza A hemagglutinin evolution. Proc. Natl. Acad. Sci. USA 2007, 104, 6283–6288. [Google Scholar] [CrossRef]
- Nakajima, S.; Nobusawa, E.; Nakajima, K. Variation in response among individuals to antigenic sites on the HA protein of human influenza virus may be responsible for the emergence of drift strains in the human population. Virology 2000, 274, 220–231. [Google Scholar]
- Deem, M.W.; Pan, K. The epitope regions of H1-subtype influenza A, with application to vaccine efficacy. Protein Eng. Des. Sel. 2009, 22, 543–546. [Google Scholar] [CrossRef]
- Tewawong, N.; Prachayangprecha, S.; Vichiwattana, P.; Korkong, S.; Klinfueng, S.; Vongpunsawad, S.; Thongmee, T.; Theamboonlers, A.; Poovorawan, Y. Assessing Antigenic Drift of Seasonal Influenza A(H3N2) and A(H1N1)pdm09 Viruses. PLoS ONE 2015, 10, e0139958. [Google Scholar]
- Cai, Z.; Zhang, T.; Wan, X.F. Concepts and applications for influenza antigenic cartography. Influenza Other Respir. Viruses 2011, 5, 204–207. [Google Scholar]
- Cai, Z.; Zhang, T.; Wan, X.F. Antigenic distance measurements for seasonal influenza vaccine selection. Vaccine 2012, 30, 448–453. [Google Scholar] [CrossRef]
- Phyu, W.W.; Saito, R. Evolutionary Dynamics of Whole-Genome Influenza A/H3N2 Viruses Isolated in Myanmar from 2015 to 2019. Viruses 2022, 14, 2414. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Earl, D.J.; Deem, M.W. Quantifying influenza vaccine efficacy and antigenic distance. Vaccine 2006, 24, 3881–3888. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Bonomo, M.E.; Deem, M.W. Predicting Influenza H3N2 Vaccine Efficacy from Evolution of the Dominant Epitope. Clin. Infect. Dis. 2018, 67, 1129–1131. [Google Scholar] [CrossRef] [PubMed]
- Wei, V.W.I.; Wong, J.Y.T.; Perera, R.; Kwok, K.O.; Fang, V.J.; Barr, I.G.; Peiris, J.S.M.; Riley, S.; Cowling, B.J. Incidence of influenza A(H3N2) virus infections in Hong Kong in a longitudinal sero-epidemiological study, 2009–2015. PLoS ONE 2018, 13, e0197504. [Google Scholar] [CrossRef] [PubMed]
- Monamele, G.C.; Vernet, M.A.; Njankouo, M.R.; Victoir, K.; Akoachere, J.F.; Anong, D.; Njouom, R. Genetic and antigenic characterization of influenza A(H3N2) in Cameroon during the 2014–2016 influenza seasons. PLoS ONE 2017, 12, e0184411. [Google Scholar]
- Lee, H.K.; Tang, J.W.; Loh, T.P.; Hurt, A.C.; Oon, L.L.; Koay, E.S. Molecular surveillance of antiviral drug resistance of influenza A/H3N2 virus in Singapore, 2009–2013. PLoS ONE 2015, 10, e0117822. [Google Scholar] [CrossRef]
- World Health Organization. Summary of Neuraminidase (NA) Amino Acid Substitutions Assessed for Their Effects on Inhibition by Neuraminidase Inhibitors (NAIs). 2023. Available online: https://www.who.int/publications/m/item/summary-of-neuraminidase-(na)-amino-acid-substitutions-associated-with-reduced-inhibition-by-neuraminidase-inhibitors-(nais) (accessed on 7 March 2023).
- Sheu, T.G.; Deyde, V.M.; Okomo-Adhiambo, M.; Garten, R.J.; Xu, X.; Bright, R.A.; Butler, E.N.; Wallis, T.R.; Klimov, A.I.; Gubareva, L.V. Surveillance for neuraminidase inhibitor resistance among human influenza A and B viruses circulating worldwide from 2004 to 2008. Antimicrob. Agents Chemother. 2008, 52, 3284–3292. [Google Scholar]
- Lackenby, A.; Besselaar, T.G.; Daniels, R.S.; Fry, A.; Gregory, V.; Gubareva, L.V.; Huang, W.; Hurt, A.C.; Leang, S.K.; Lee, R.T.C.; et al. Global update on the susceptibility of human influenza viruses to neuraminidase inhibitors and status of novel antivirals, 2016–2017. Antiviral. Res. 2018, 157, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Monto, A.S.; McKimm-Breschkin, J.L.; Macken, C.; Hampson, A.W.; Hay, A.; Klimov, A.; Tashiro, M.; Webster, R.G.; Aymard, M.; Hayden, F.G.; et al. Detection of influenza viruses resistant to neuraminidase inhibitors in global surveillance during the first 3 years of their use. Antimicrob. Agents Chemother. 2006, 50, 2395–2402. [Google Scholar] [CrossRef]
- Dapat, C.; Kondo, H.; Dapat, I.C.; Baranovich, T.; Suzuki, Y.; Shobugawa, Y.; Saito, K.; Saito, R.; Suzuki, H. Neuraminidase inhibitor susceptibility profile of pandemic and seasonal influenza viruses during the 2009–2010 and 2010–2011 influenza seasons in Japan. Antiviral. Res. 2013, 99, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Rimmelzwaan, G.F.; Kreijtz, J.H.; Bodewes, R.; Fouchier, R.A.; Osterhaus, A.D. Influenza virus CTL epitopes, remarkably conserved and remarkably variable. Vaccine 2009, 27, 6363–6365. [Google Scholar] [CrossRef]
- Simon, B.; Pichon, M. Whole Genome Sequencing of A(H3N2) Influenza Viruses Reveals Variants Associated with Severity during the 2016–2017 Season. Viruses 2019, 11, 108. [Google Scholar] [CrossRef]
- Wei, D.; Yu, D.M.; Wang, M.J.; Zhang, D.H.; Cheng, Q.J.; Qu, J.M.; Zhang, X.X. Genome-wide characterization of the seasonal H3N2 virus in Shanghai reveals natural temperature-sensitive strains conferred by the I668V mutation in the PA subunit. Emerg. Microbes. Infect. 2018, 7, 171. [Google Scholar] [PubMed]
- Chen, W.; Calvo, P.A.; Malide, D.; Gibbs, J.; Schubert, U.; Bacik, I.; Basta, S.; O’Neill, R.; Schickli, J.; Palese, P.; et al. A novel influenza A virus mitochondrial protein that induces cell death. Nat. Med. 2001, 7, 1306–1312. [Google Scholar] [PubMed]
- McAuley, J.L.; Hornung, F.; Boyd, K.L.; Smith, A.M.; McKeon, R.; Bennink, J.; Yewdell, J.W.; McCullers, J.A. Expression of the 1918 influenza A virus PB1-F2 enhances the pathogenesis of viral and secondary bacterial pneumonia. Cell Host Microbe 2007, 2, 240–249. [Google Scholar] [CrossRef]
- Mazur, I.; Anhlan, D.; Mitzner, D.; Wixler, L.; Schubert, U.; Ludwig, S. The proapoptotic influenza A virus protein PB1-F2 regulates viral polymerase activity by interaction with the PB1 protein. Cell. Microbiol. 2008, 10, 1140–1152. [Google Scholar] [PubMed]
- Patel, M.C.; Chesnokov, A.; Jones, J.; Mishin, V.P.; De La Cruz, J.A.; Nguyen, H.T.; Zanders, N.; Wentworth, D.E.; Davis, T.C.; Gubareva, L.V. Susceptibility of widely diverse influenza a viruses to PB2 polymerase inhibitor pimodivir. Antiviral. Res. 2021, 188, 105035. [Google Scholar] [CrossRef]
- Pond, S.L.; Frost, S.D. Datamonkey: Rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 2005, 21, 2531–2533. [Google Scholar]
- Suntronwong, N.; Klinfueng, S.; Korkong, S.; Vichaiwattana, P.; Thongmee, T.; Vongpunsawad, S.; Poovorawan, Y. Characterizing genetic and antigenic divergence from vaccine strain of influenza A and B viruses circulating in Thailand, 2017–2020. Sci. Rep. 2021, 11, 735. [Google Scholar] [CrossRef] [PubMed]
- Suntronwong, N.; Klinfueng, S.; Vichiwattana, P.; Korkong, S.; Thongmee, T.; Vongpunsawad, S.; Poovorawan, Y. Genetic and antigenic divergence in the influenza A(H3N2) virus circulating between 2016 and 2017 in Thailand. PLoS ONE 2017, 12, e0189511. [Google Scholar]
- Kawakami, C.; Yamayoshi, S.; Akimoto, M.; Nakamura, K.; Miura, H.; Fujisaki, S.D.; Pattinson, J.; Shimizu, K.; Ozawa, H.; Momoki, T.; et al. Genetic and antigenic characterisation of influenza A(H3N2) viruses isolated in Yokohama during the 2016/17 and 2017/18 influenza seasons. Eurosurveillance 2019, 24, 1800467. [Google Scholar]
- Zhang, Z.; Li, S.; Zhu, X.; Hou, J.; Zhang, H.; Zhao, B.; Tian, Z. Increased genetic variation of A(H3N2) virus from influenza surveillance at the end of the 2016/2017 season for Shanghai port, China. Sci. Rep. 2022, 12, 17089. [Google Scholar] [CrossRef]
- Hernández-Hernández, V.A.; Higuera-Iglesias, A.L.; Palma-Cortes, G.; Tapia-Trejo, D.; Ávila-Ríos, S.; González-Fernández, R.R.; Pérez-Moreno, L.; Zuñiga-Ramos, J.A.; Guadarrama-Pérez, C.; Sandoval-Gutiérrez, J.L.; et al. A(H3N2) antigenic variation of influenza is associated with low vaccine efficacy in the early 2018 influenza season in Mexico City. Int. J. Infect. Dis. 2022, 125, 114–119. [Google Scholar]
- Galli, C.; Pellegrinelli, L.; Giardina, F.; Ferrari, G.; Renteria, S.C.U.; Novazzi, F.; Masi, E.; Pagani, E.; Piccirilli, G.; Mauro, M.V.; et al. On the lookout for influenza viruses in Italy during the 2021–2022 season: Along came A(H3N2) viruses with a new phylogenetic makeup of their hemagglutinin. Virus. Res. 2023, 324, 199033. [Google Scholar] [CrossRef]
- Pendrey, C.G.; Strachan, J.; Peck, H.; Aziz, A.; Moselen, J.; Moss, R.; Rahaman, M.R.; Barr, I.G.; Subbarao, K.; Sullivan, S.G. The re-emergence of influenza following the COVID-19 pandemic in Victoria, Australia, 2021 to 2022. EuroSurveill 2023, 28, 2300118. [Google Scholar] [CrossRef]
- Chon, I.; Saito, R. Whole-Genome Analysis of Influenza A(H3N2) and B/Victoria Viruses Detected in Myanmar during the COVID-19 Pandemic in 2021. Viruses 2023, 15, 583. [Google Scholar] [PubMed]
- Ferguson, N.M.; Galvani, A.P.; Bush, R.M. Ecological and immunological determinants of influenza evolution. Nature 2003, 422, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Wilson, I.A.; Cox, N.J. Structural basis of immune recognition of influenza virus hemagglutinin. Annu. Rev. Immunol. 1990, 8, 737–771. [Google Scholar] [PubMed]
- Njifon, H.L.M.; Monamele, C.G.; Vernet, M.A.; Njankouo, M.R.; Deweerdt, L.; Nono, R.; Kenmoe, S.; Mbacham, W.; Njouom, R. Genetic diversity of influenza A(H3N2) viruses in Northern Cameroon during the 2014–2016 influenza seasons. J. Med. Virol. 2019, 91, 1400–1407. [Google Scholar] [CrossRef]
- Potdar, V.A.; Dakhave, M.R.; Kulkarni, P.B.; Tikhe, S.A.; Broor, S.; Gunashekaran, P.; Chawla-Sarkar, M.; Abraham, A.; Bishwas, D.; Patil, K.N.; et al. Antiviral drug profile of human influenza A & B viruses circulating in India: 2004–2011. Indian J. Med. Res. 2014, 140, 244–251. [Google Scholar]
- Zaraket, H.; Kondo, H.; Hibino, A.; Yagami, R.; Odagiri, T.; Takemae, N.; Tsunekuni, R.; Saito, T.; Myint, Y.Y.; Kyaw, Y.; et al. Full Genome Characterization of Human Influenza A/H3N2 Isolates from Asian Countries Reveals a Rare Amantadine Resistance-Conferring Mutation and Novel PB1-F2 Polymorphisms. Front. Microbiol. 2016, 7, 262. [Google Scholar] [CrossRef]
- Barreiro, L.B.; Quintana-Murci, L. From evolutionary genetics to human immunology: How selection shapes host defence genes. Nat. Rev. Genet. 2010, 11, 17–30. [Google Scholar] [CrossRef]
- Chen, R.; Holmes, E.C. The evolutionary dynamics of human influenza B virus. J. Mol. Evol. 2008, 66, 655–663. [Google Scholar]
- Westgeest, K.B.; Russell, C.A.; Lin, X.; Spronken, M.I.; Bestebroer, T.M.; Bahl, J.; van Beek, R.; Skepner, E.; Halpin, R.A.; de Jong, J.C.; et al. Genomewide analysis of reassortment and evolution of human influenza A(H3N2) viruses circulating between 1968 and 2011. J. Virol. 2014, 88, 2844–2857. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Coding Region | SLAC (Single-Likelihood Ancestor Counting) | MEME (Mixed Effects Model of Evolution) | FEL (Fixed Effects Likelihood) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| dN/dS | Position | p-Value | dN/dS | Position | p-Value | dN/dS | Position | p-Value | |
| PB2 | 0.079 | N/D | N/D | 0.075 | 255 | 0.090 | 0.079 | 255 | 0.065 |
| 451 | 0.000 | ||||||||
| PB1 | 0.084 | N/D | N/D | 0.078 | N/D | N/D | 0.084 | N/D | N/D |
| PA | 0.088 | N/D | N/D | 0.081 | N/D | N/D | 0.088 | N/D | N/D |
| HA | 0.258 | 176 | 0.000 | 0.228 | 147 | 0.000 | 0.258 | 176 | 0.000 |
| 277 | 0.023 | 160 | 0.100 | 202 | 0.075 | ||||
| 176 | 0.000 | 277 | 0.011 | ||||||
| 202 | 0.040 | ||||||||
| 277 | 0.020 | ||||||||
| 363 | 0.040 | ||||||||
| NP | 0.068 | N/D | N/D | 0.063 | N/D | N/D | 0.063 | N/D | N/D |
| NA | 0.268 | 346 | 0.099 | 0.235 | 140 | 0.080 | 0.268 | 140 | 0.060 |
| 380 | 0.091 | 143 | 0.050 | 143 | 0.036 | ||||
| 380 | 0.070 | 346 | 0.087 | ||||||
| 380 | 0.053 | ||||||||
| M1 | 0.022 | N/D | N/D | 0.020 | N/D | N/D | 0.020 | N/D | N/D |
| M2 | 0.514 | N/D | N/D | 0.483 | 25 | 0.070 | 0.483 | 25 | 0.053 |
| NS1 | 0.455 | N/D | N/D | 0.445 | N/D | N/D | 0.445 | N/D | N/D |
| NS2 | 0.260 | N/D | N/D | 0.243 | N/D | N/D | 0.243 | N/D | N/D |
| Year (N) | Vaccine Strain | No. of Strains | Dominant Epitope | No. of Mutations | Residue Differences | Pepitope | Vaccine Efficacy (%) |
|---|---|---|---|---|---|---|---|
| 2017 (N = 34) | A/Hong Kong/4801/2014 | 16 | A | 2 | T131K, R142K | 0.1053 | 21.00 |
| 2018 (N = 18) | A/Singapore/INFIMH-16-0019/2016 | 10 | A | 1 | T135K/N, | 0.0526 | 34.00 |
| 1 | A | 2 | S144K, R150K | 0.1053 | 21.00 | ||
| 2019 (N = 79) | A/Kansas/14/2017 | 79 | E | 3 | E62G, N91S, K92R | 0.1364 | 13.32 |
| 2020 (N = 30) | A/Hong Kong/45/2019 | 30 | C | 1 | N312S | 0.0370 | 37.85 |
| 2021 (N = 8) | A/Cambodia/e0826360/2020 | 8 | A | 4 | K131T, T135K, S137F, A138S | 0.2105 | −5.00 |
| 2022 (N = 84) | A/Darwin/6/2021 | 84 | B | 5 | S156H, N159Y, D186S, N190D, S198P | 0.2381 | −11.81 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, M.; Zhou, J.; Ni, R.; Zhao, X.; Chen, Y.; Sun, Y.; Liu, Z.; Han, X.; Luo, C.; Fu, X.; et al. Genomic Analyses Uncover Evolutionary Features of Influenza A/H3N2 Viruses in Yunnan Province, China, from 2017 to 2022. Viruses 2024, 16, 138. https://doi.org/10.3390/v16010138
Zhang M, Zhou J, Ni R, Zhao X, Chen Y, Sun Y, Liu Z, Han X, Luo C, Fu X, et al. Genomic Analyses Uncover Evolutionary Features of Influenza A/H3N2 Viruses in Yunnan Province, China, from 2017 to 2022. Viruses. 2024; 16(1):138. https://doi.org/10.3390/v16010138
Chicago/Turabian StyleZhang, Meiling, Jienan Zhou, Ruize Ni, Xiaonan Zhao, Yaoyao Chen, Yanhong Sun, Zhaosheng Liu, Xiaoyu Han, Chunrui Luo, Xiaoqing Fu, and et al. 2024. "Genomic Analyses Uncover Evolutionary Features of Influenza A/H3N2 Viruses in Yunnan Province, China, from 2017 to 2022" Viruses 16, no. 1: 138. https://doi.org/10.3390/v16010138
APA StyleZhang, M., Zhou, J., Ni, R., Zhao, X., Chen, Y., Sun, Y., Liu, Z., Han, X., Luo, C., Fu, X., & Shao, Y. (2024). Genomic Analyses Uncover Evolutionary Features of Influenza A/H3N2 Viruses in Yunnan Province, China, from 2017 to 2022. Viruses, 16(1), 138. https://doi.org/10.3390/v16010138