
Comprehensive Characterization of Viral Diversity of Female Mosquitoes in Madagascar

 , , , ,
, , , ,  , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
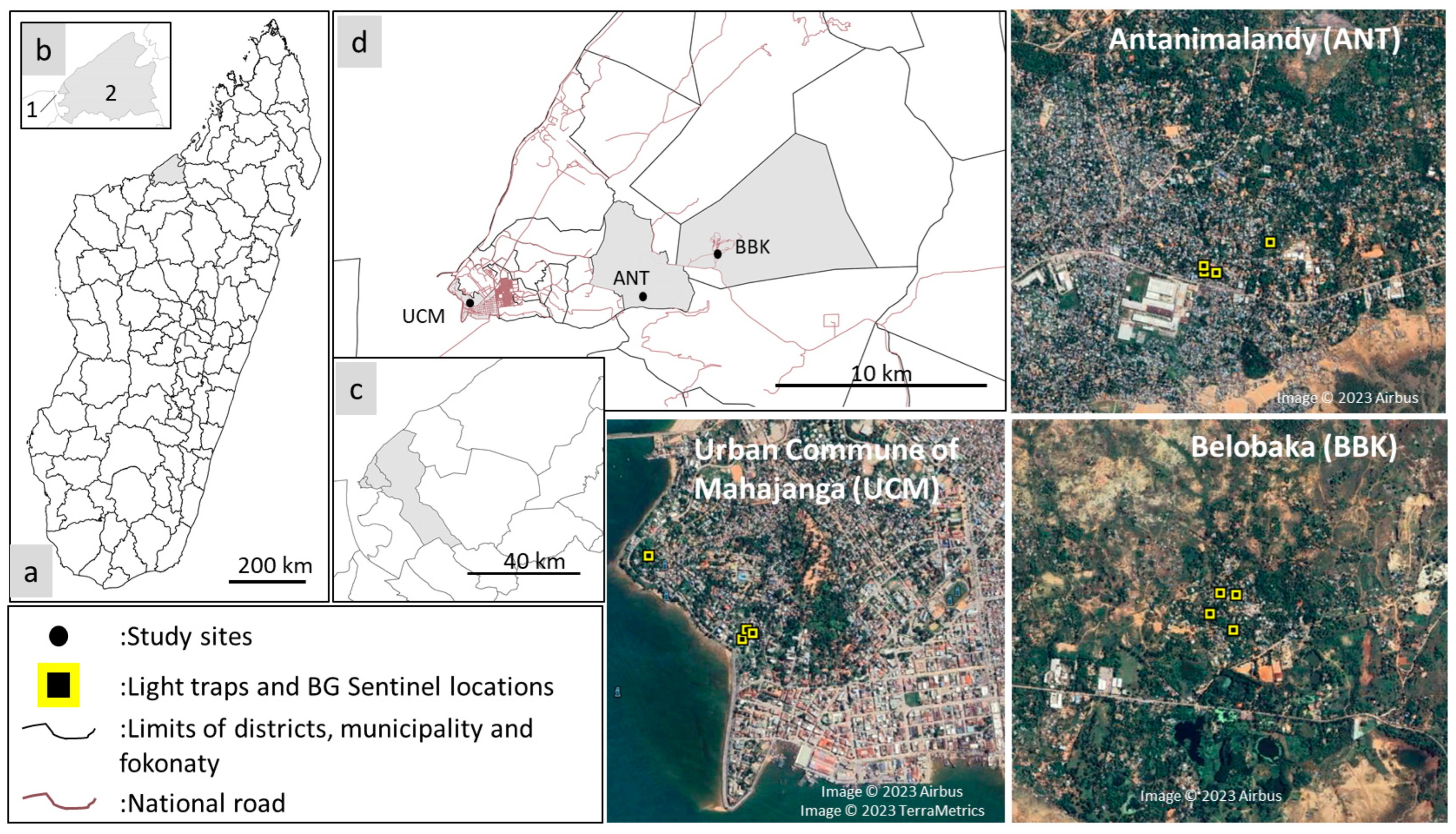
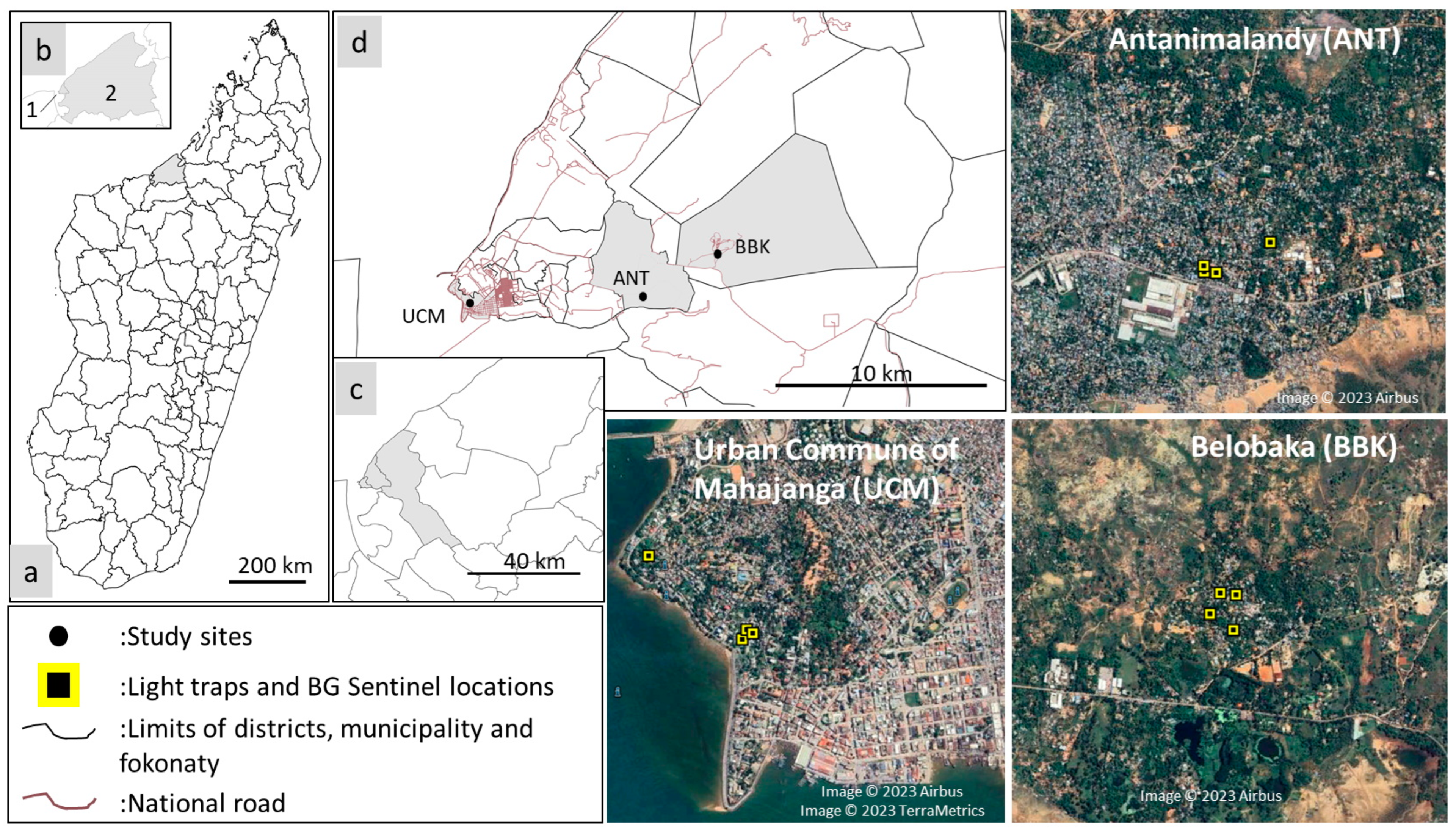
2.1. Study Sites
2.2. Mosquito Sampling, Identification, and Storage
2.3. RNA Extraction, NGS Libraries Construction, and Sequencing
2.4. Virus Taxonomic Assignation
2.5. Statistical Analysis
2.6. Phylogenetic Analysis
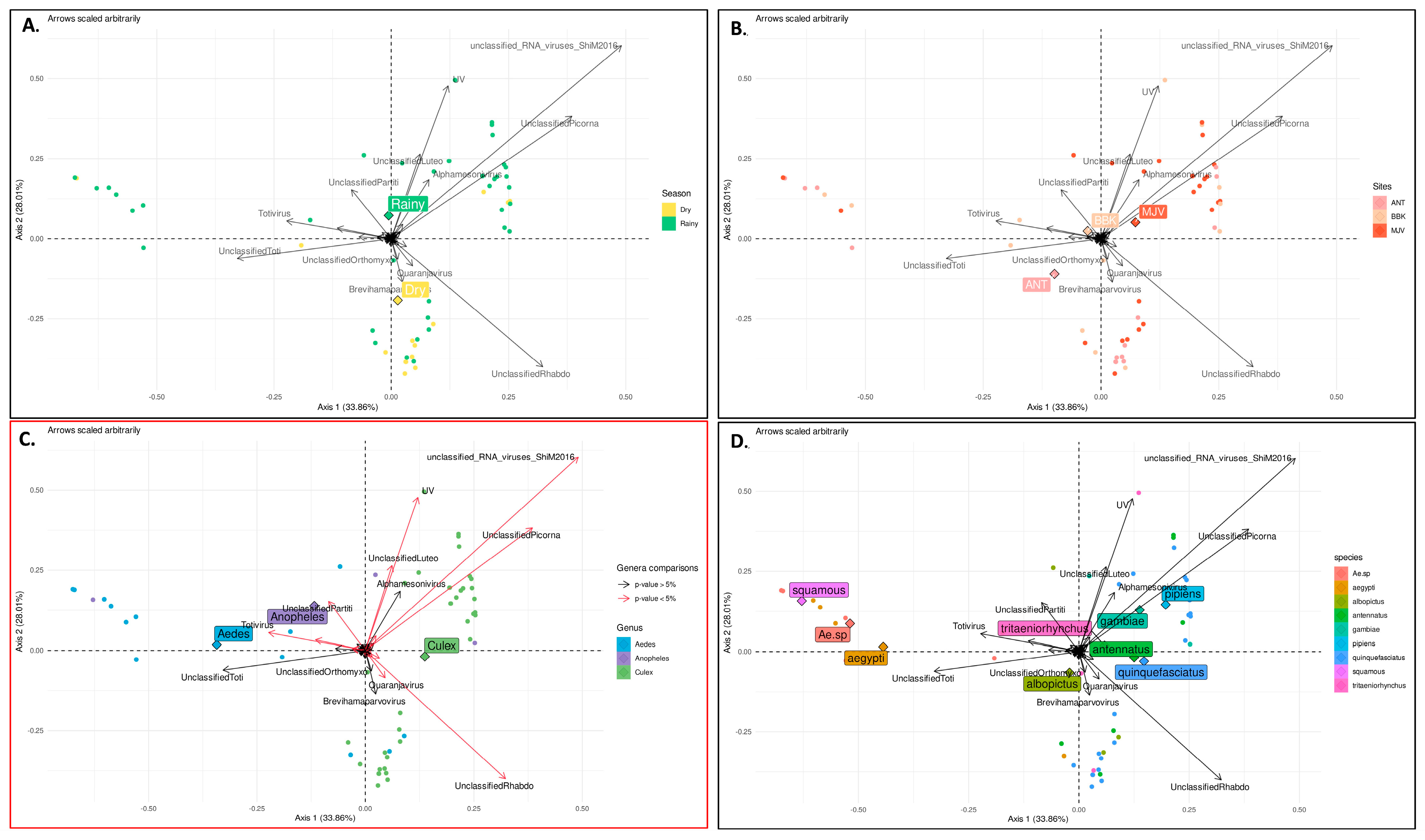
3. Results
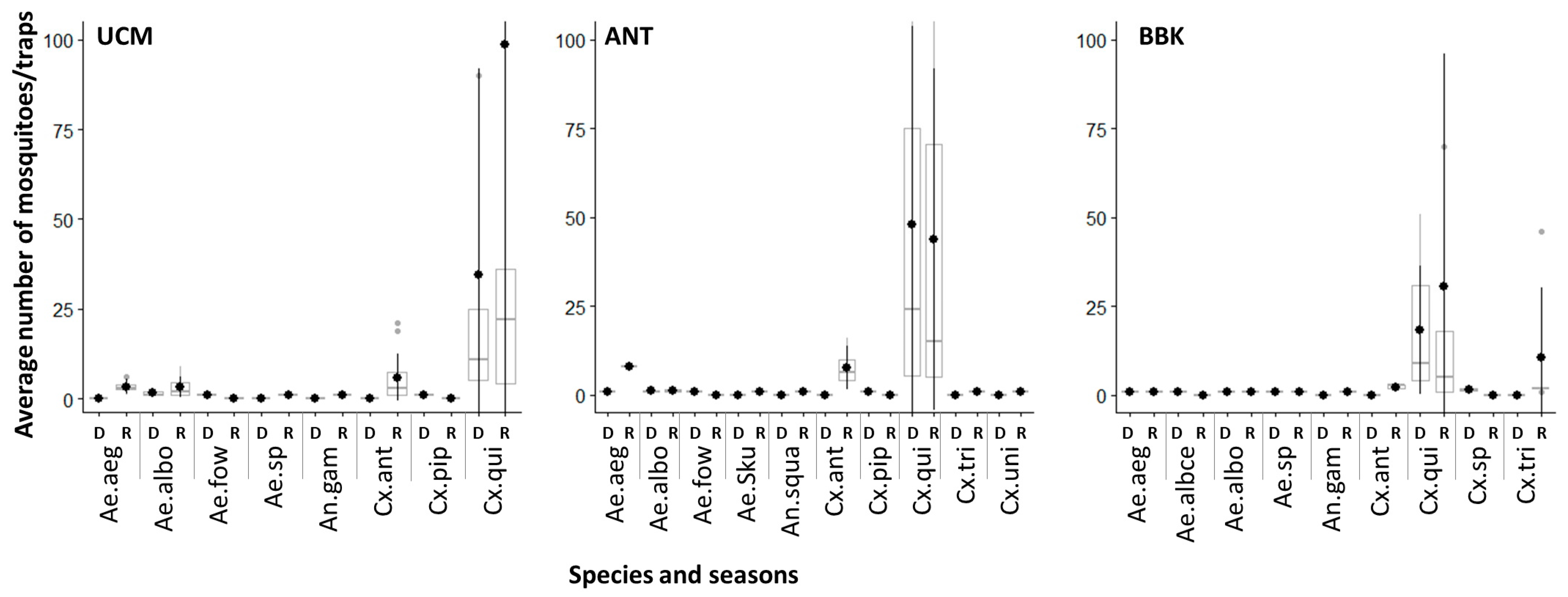
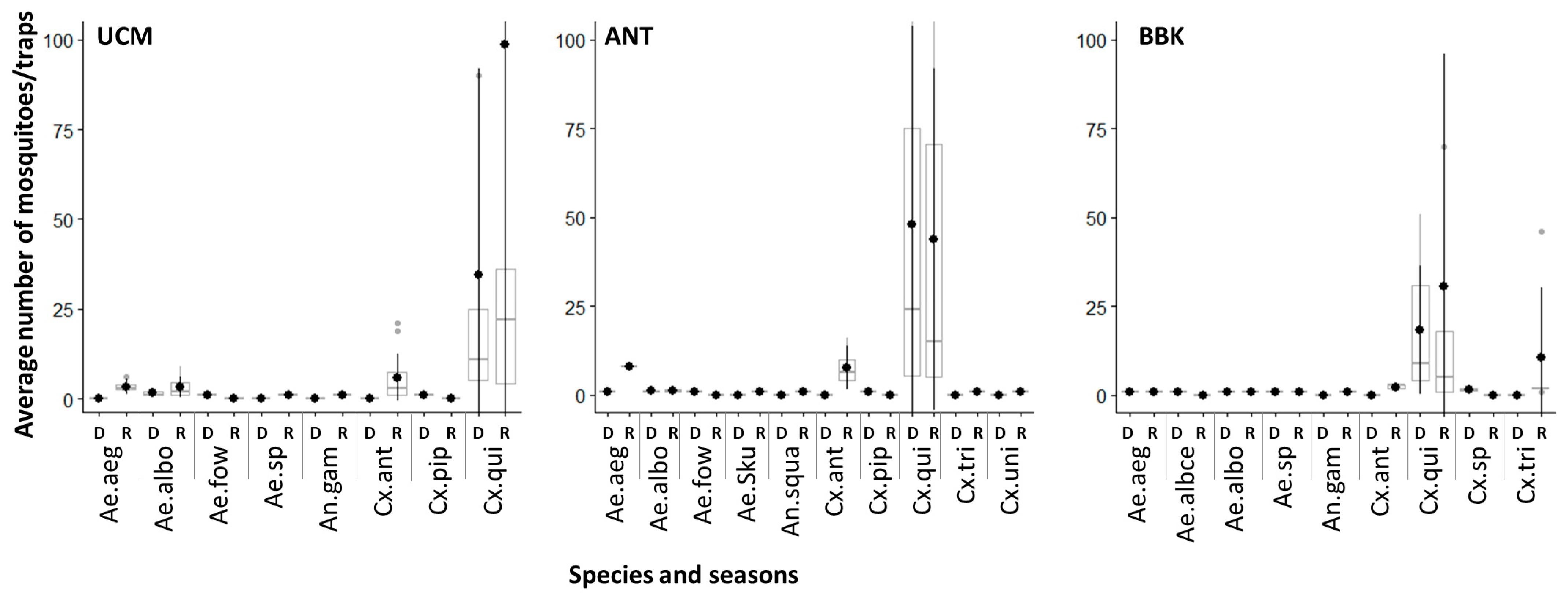
3.1. Mosquito Diversity across Sites and Seasons
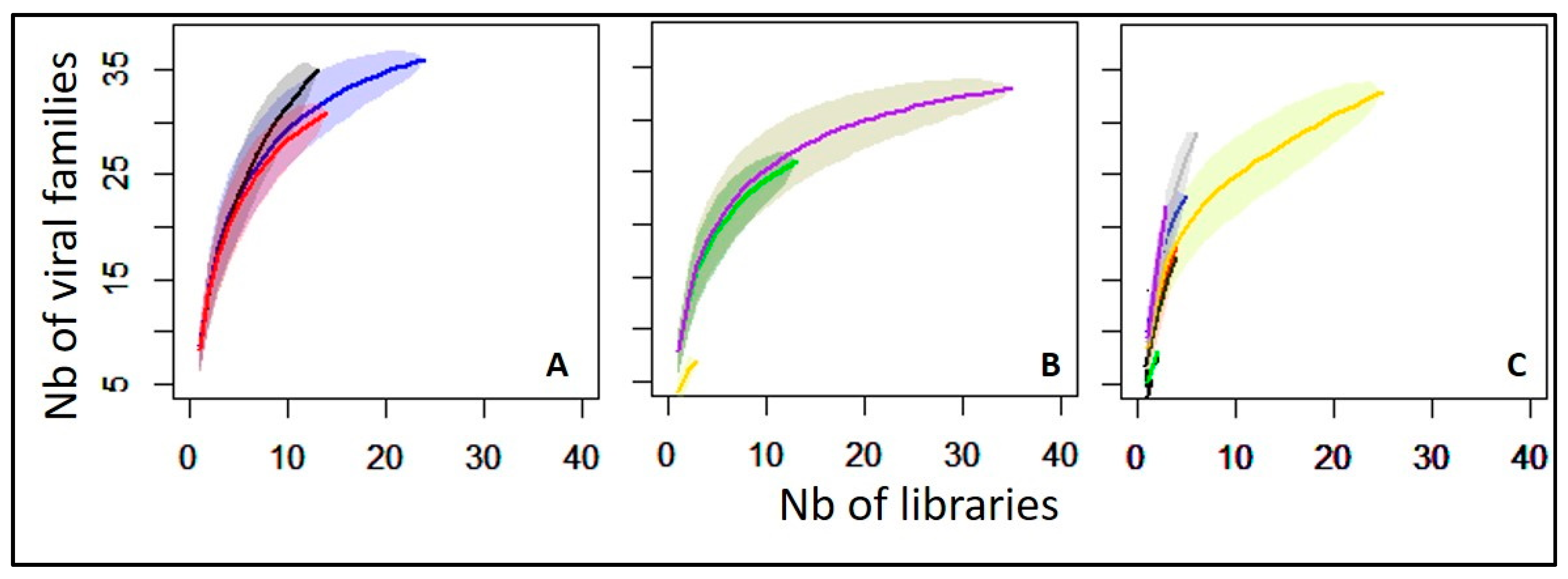
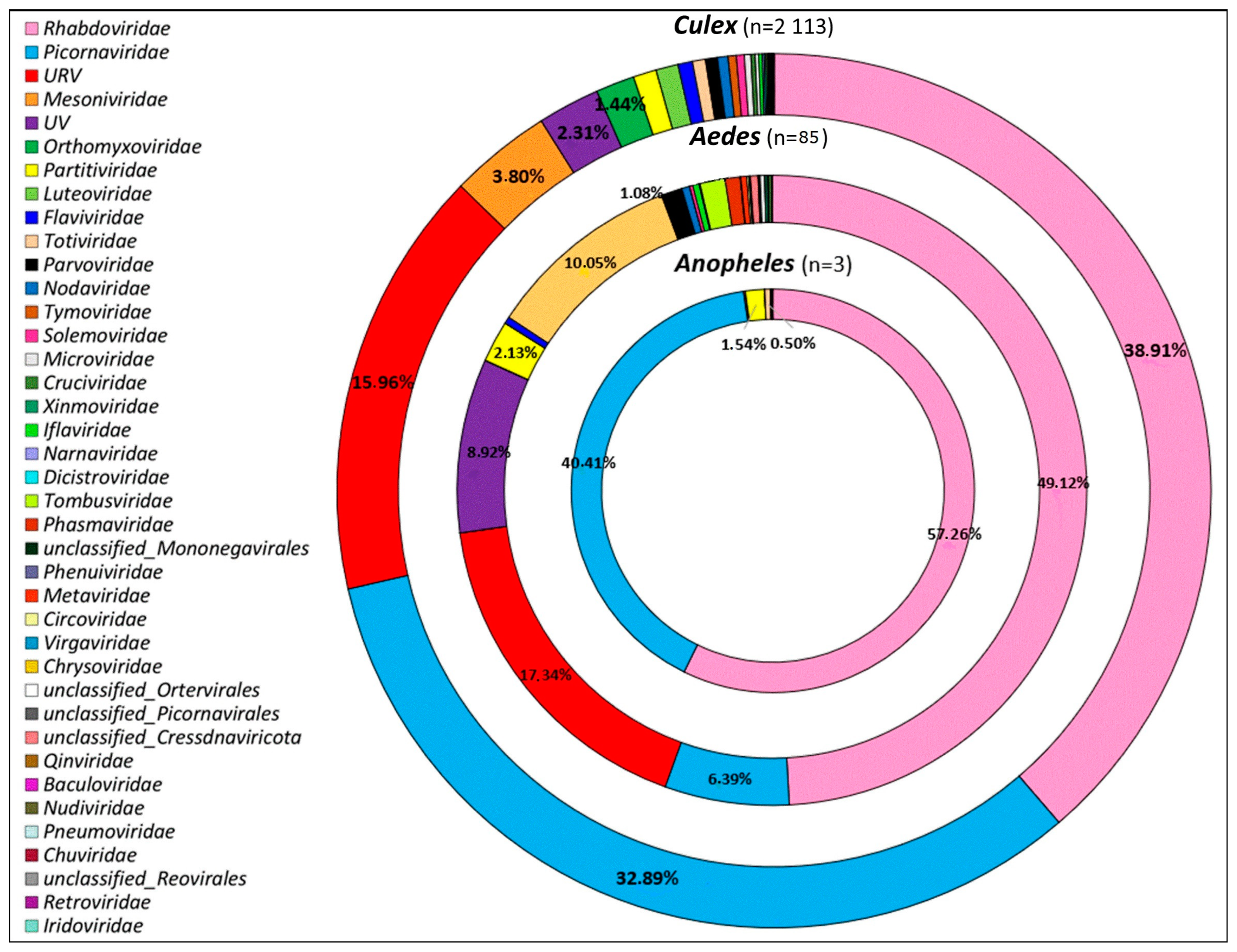
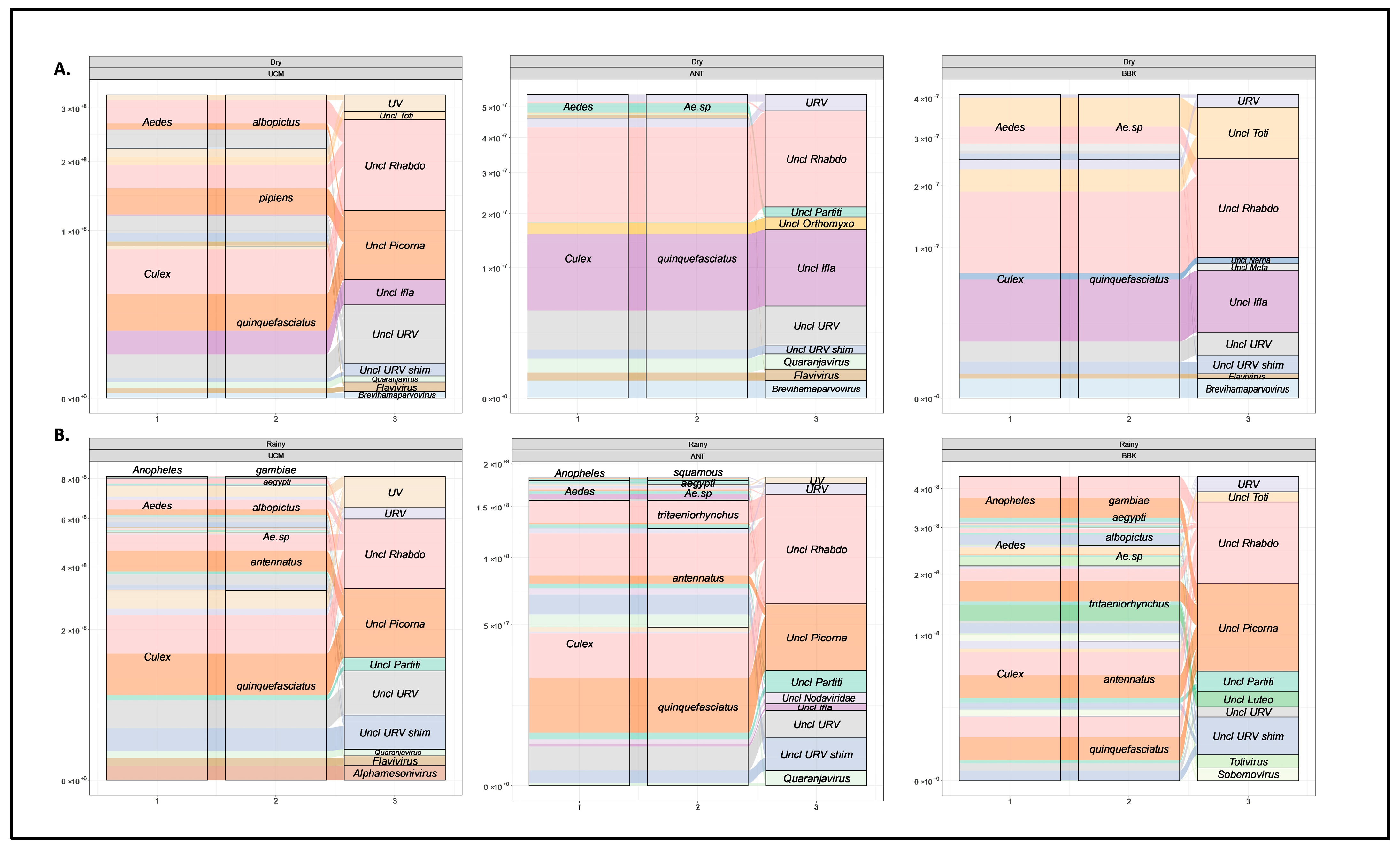
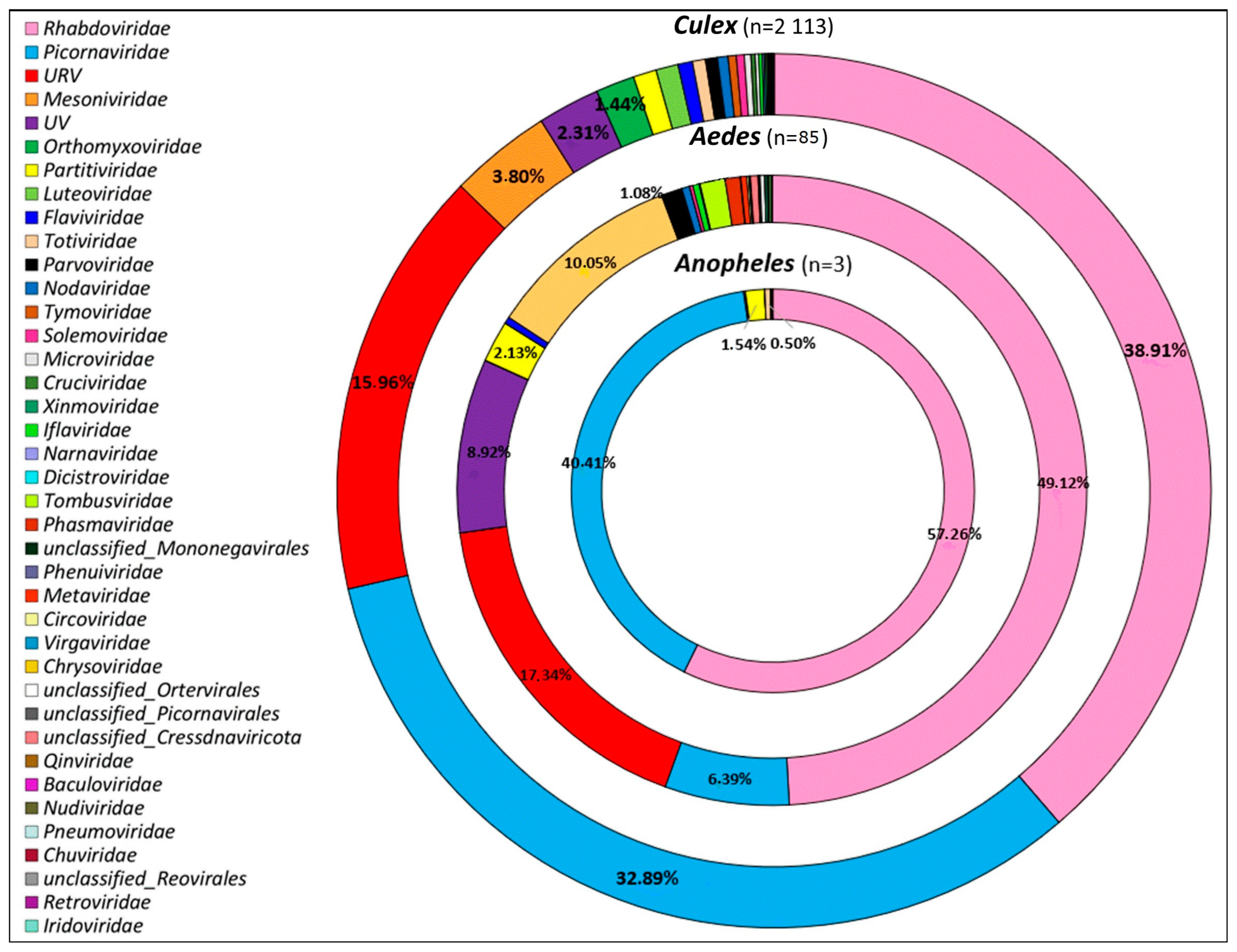
3.2. Viral Families Diversity by Site, Mosquitoes Genus, and Species
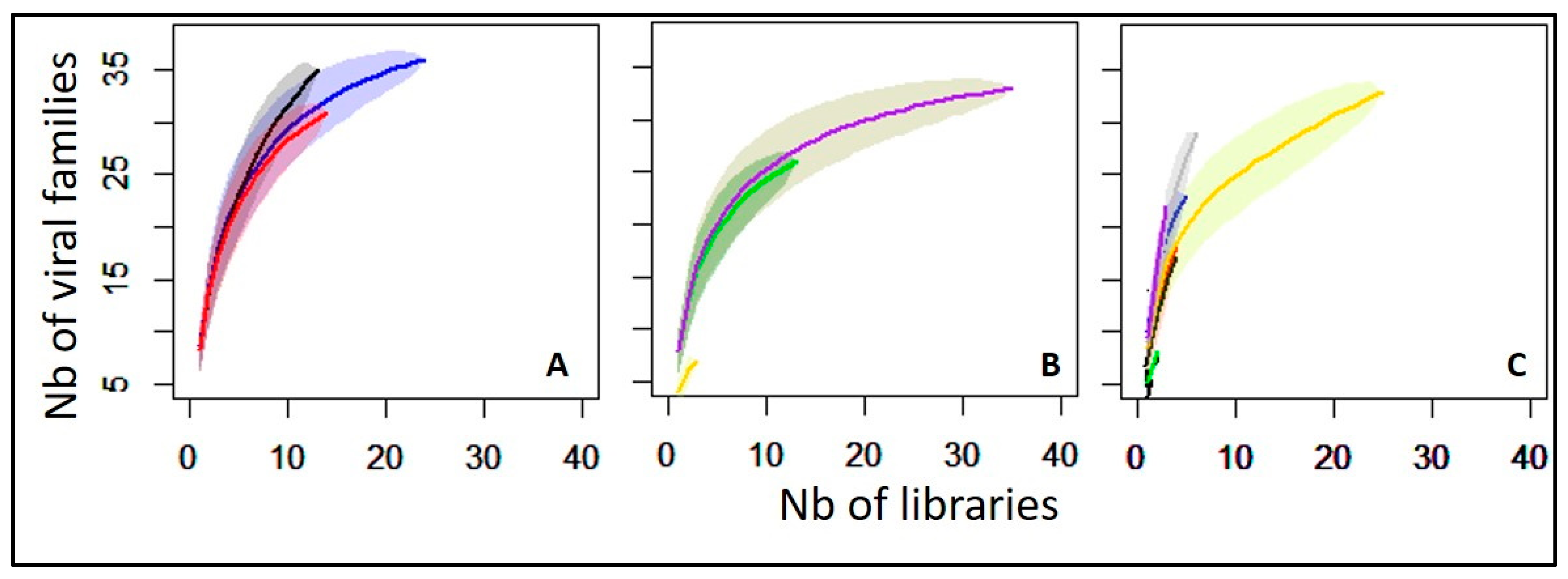
3.3. Overview of Virus Diversity Carried by Mosquitoes
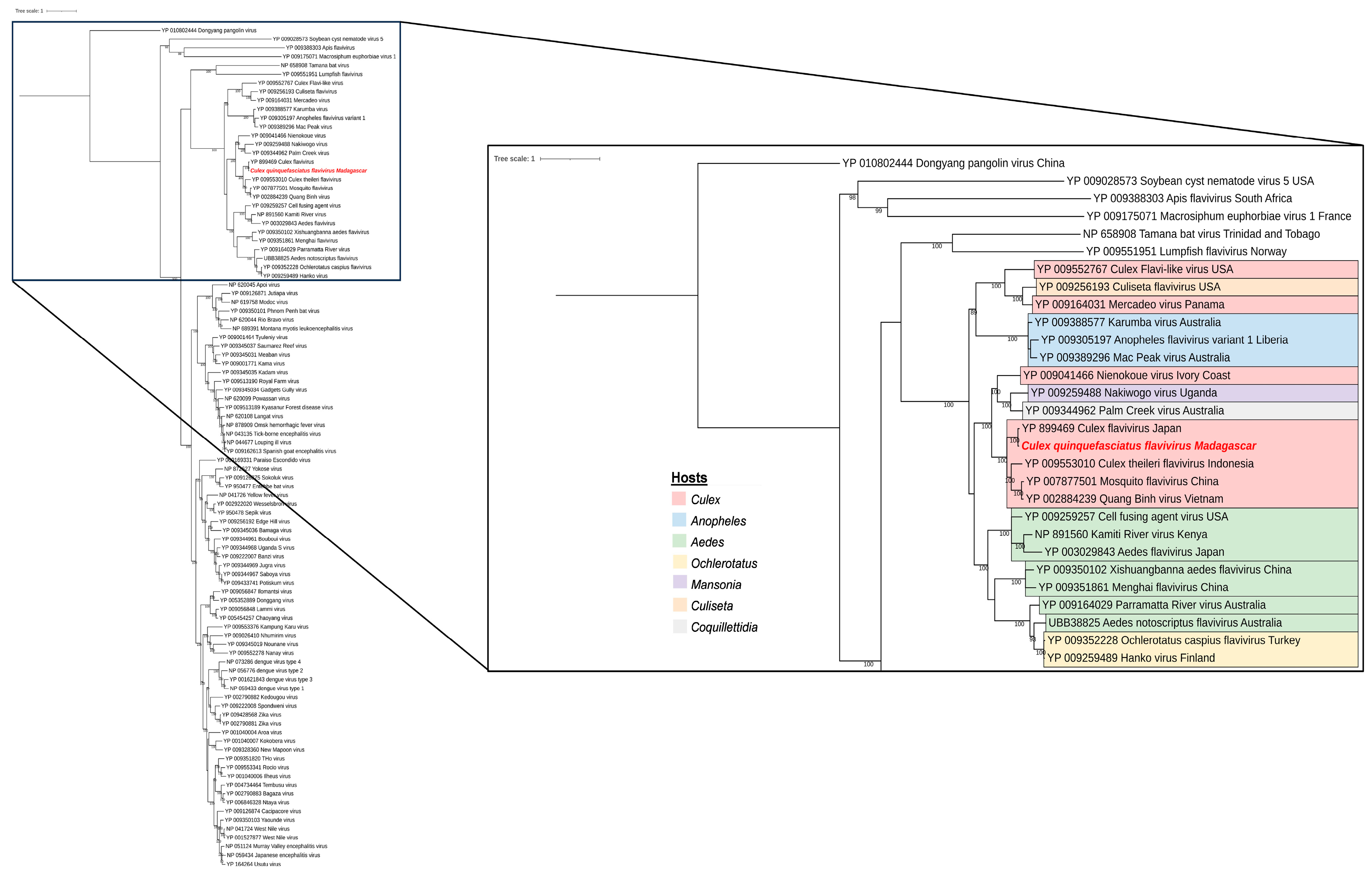
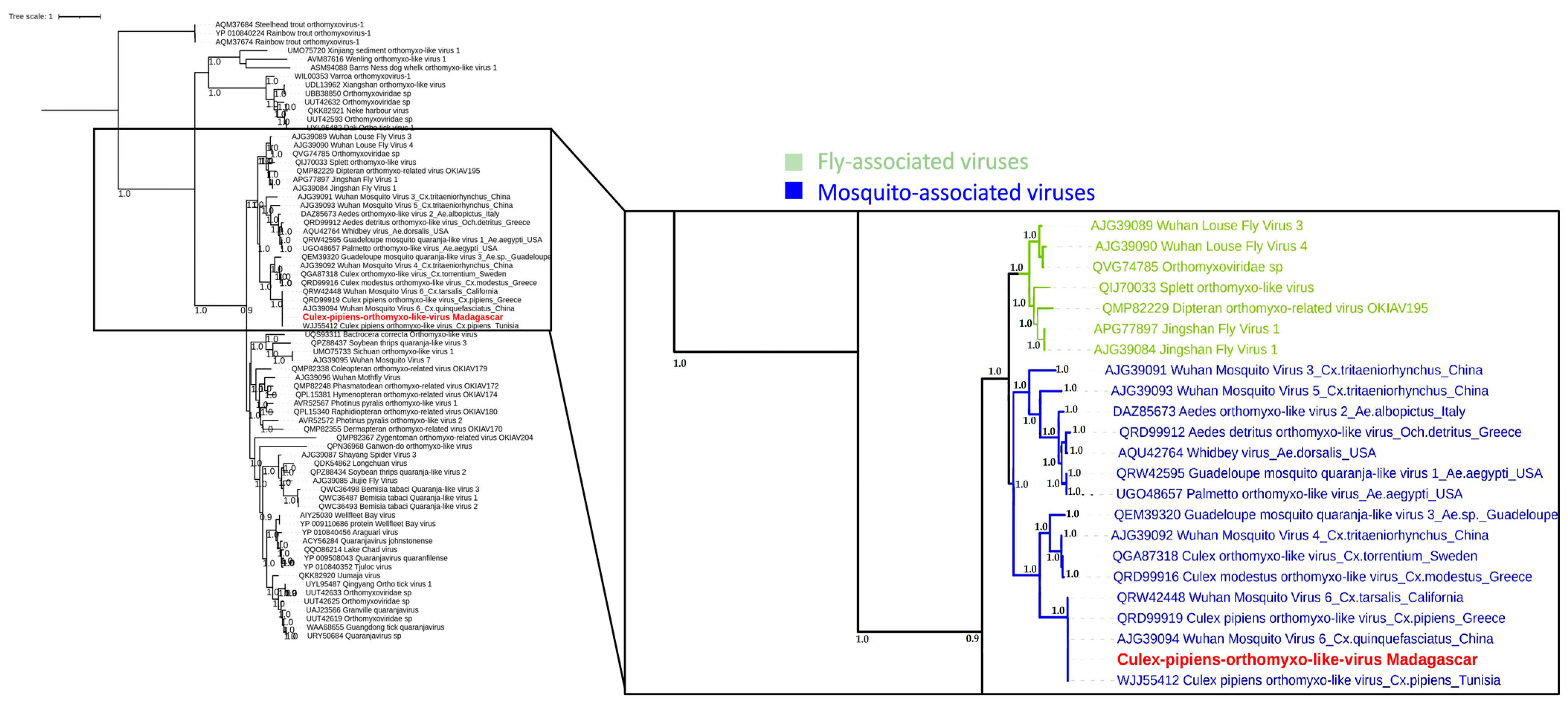
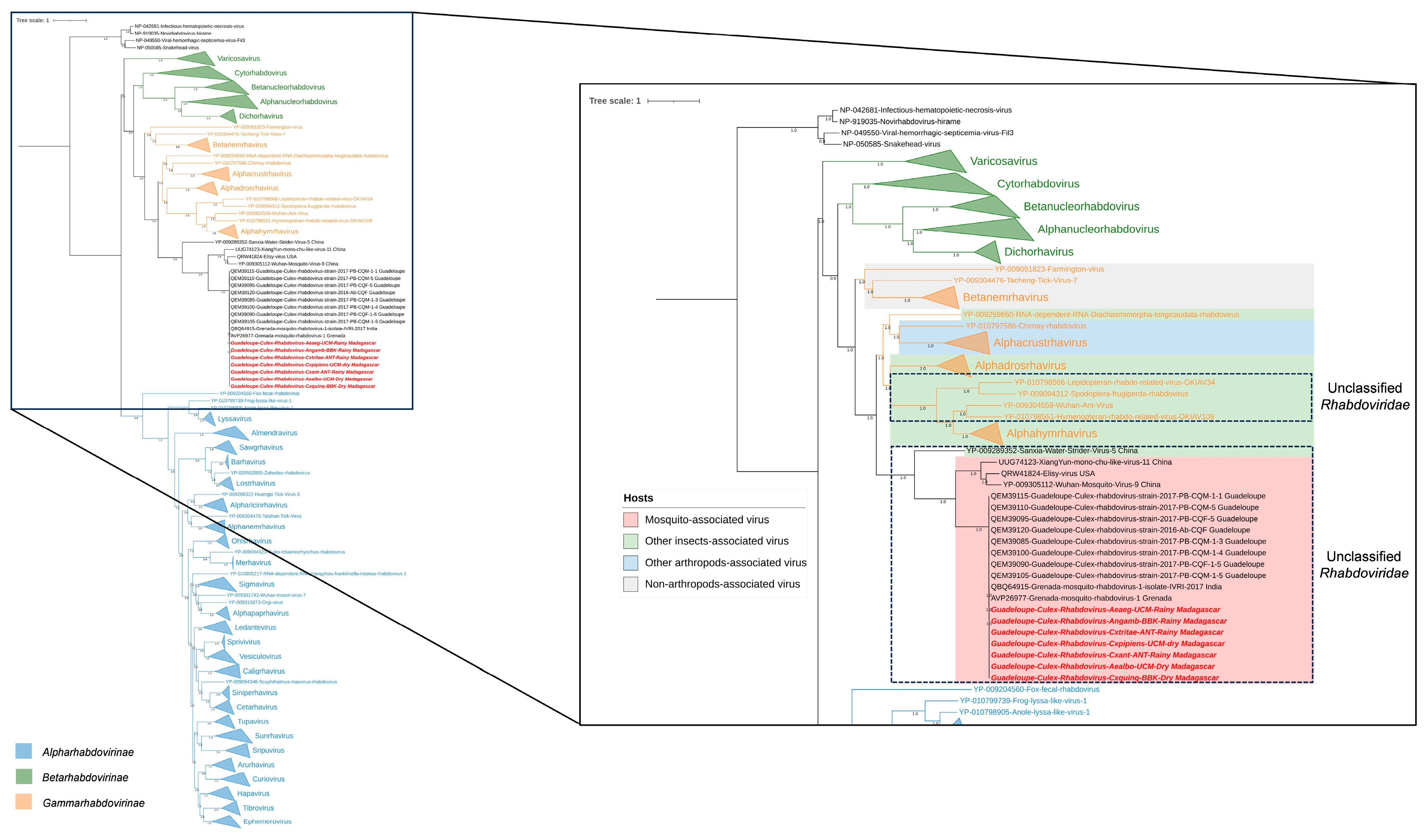
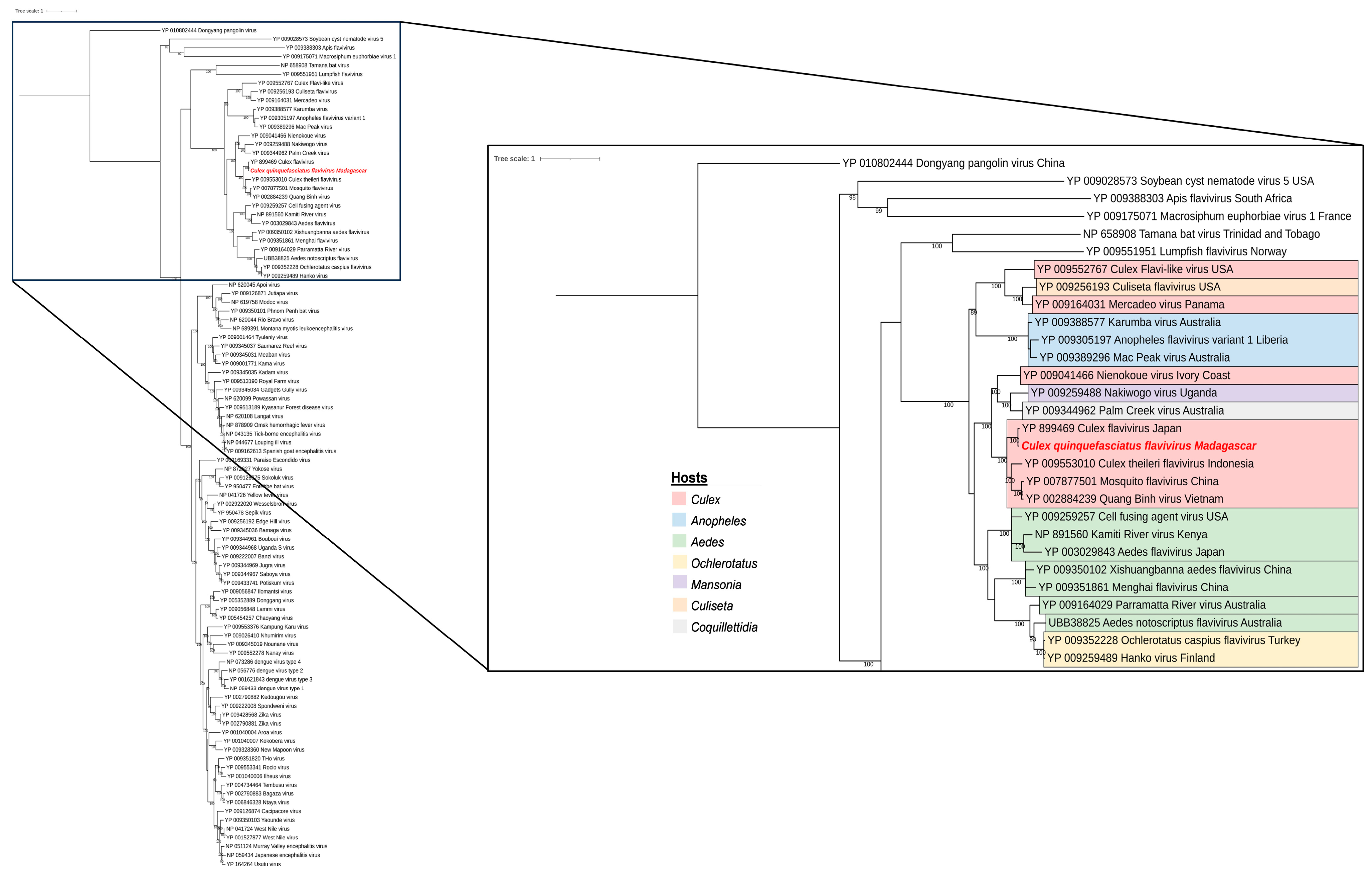
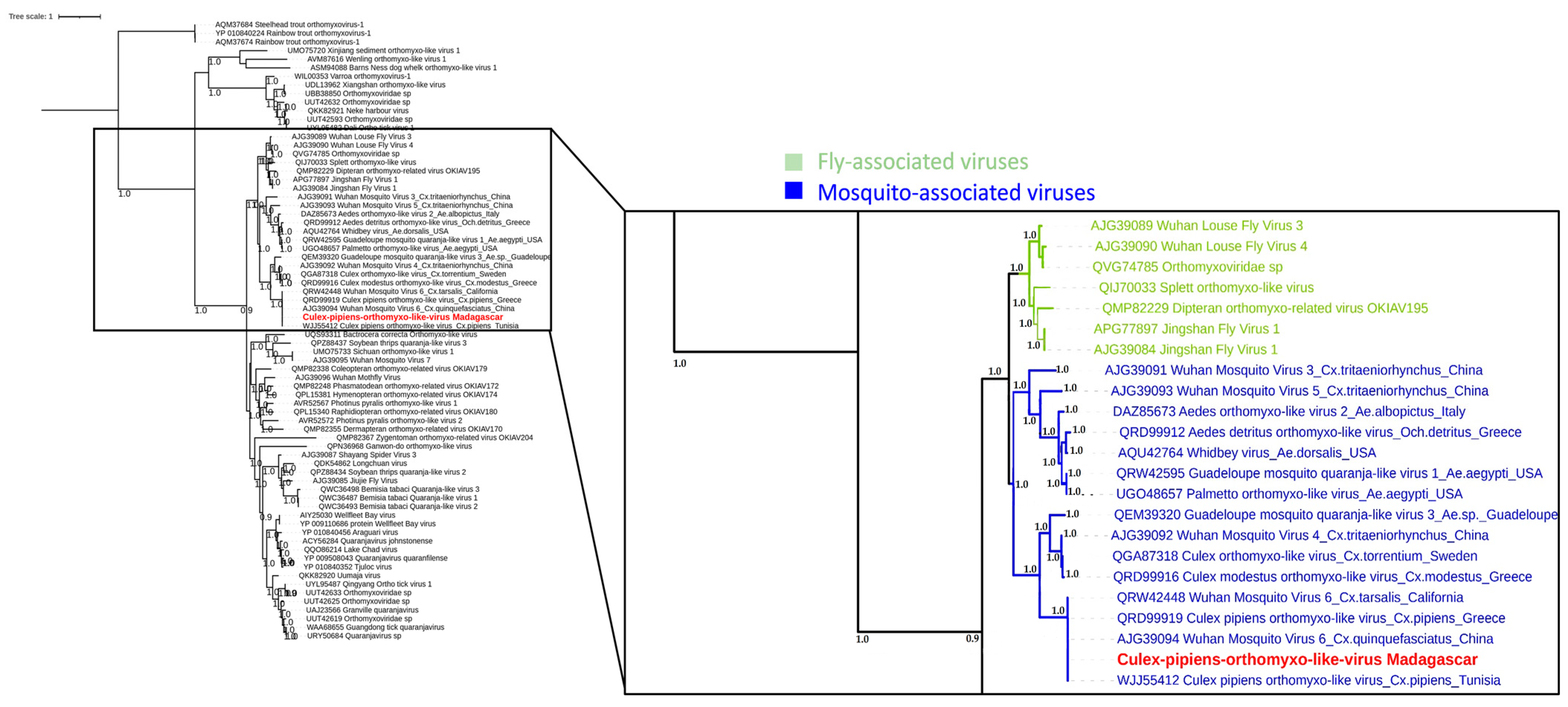
3.4. Phylogenetic Analysis of Selected Viral Species
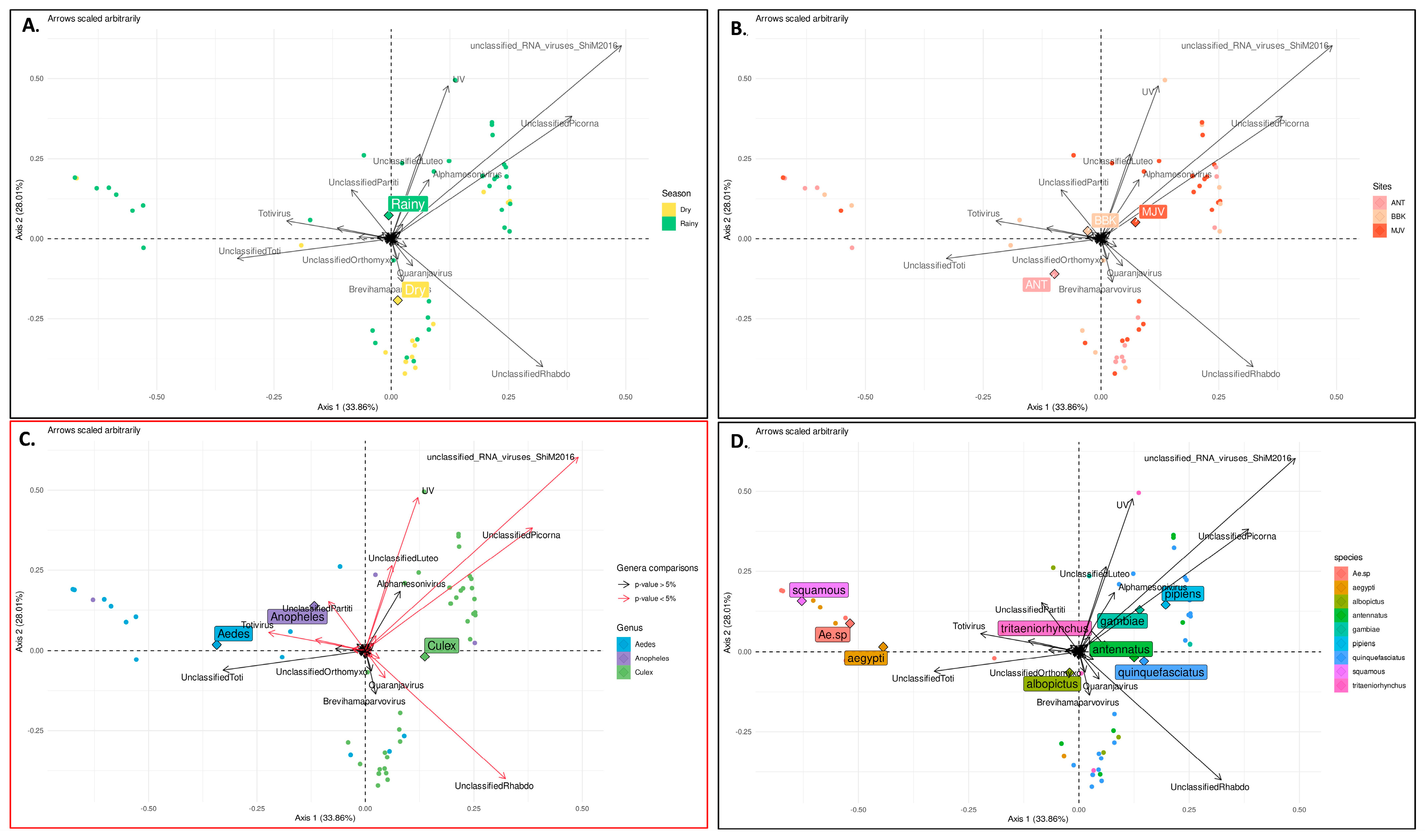
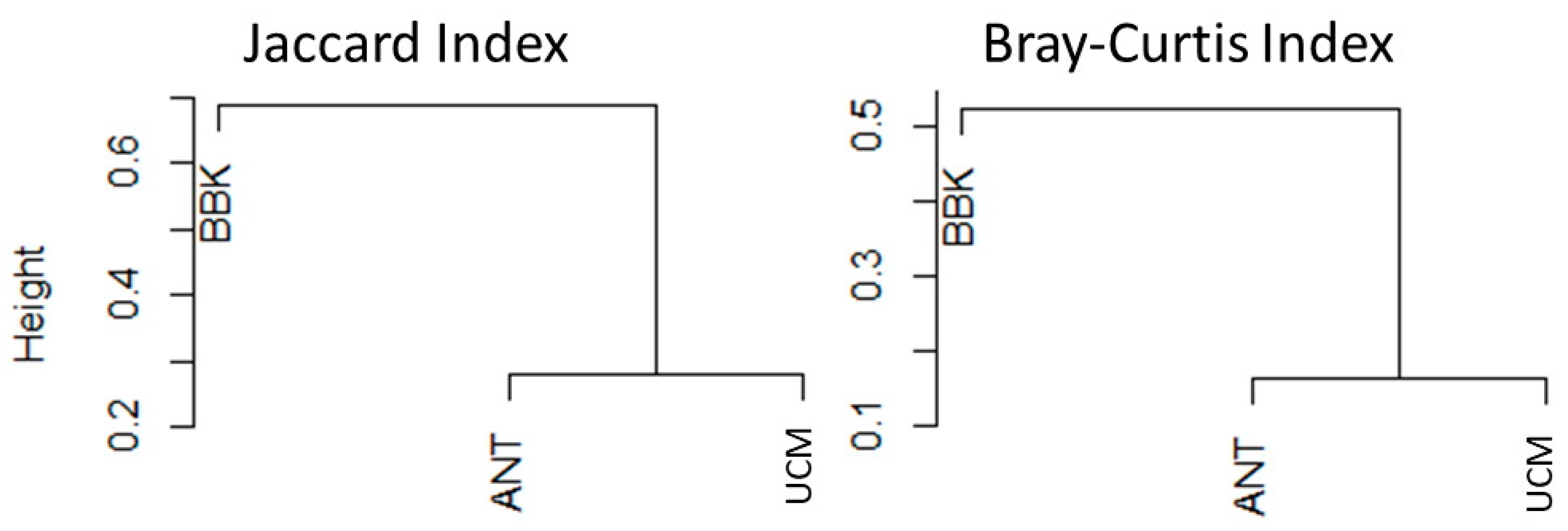
3.5. Factors That Shape Mosquito Viral Communities in Mahajanga
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Molaei, G.; Andreadis, T.G.; Armstrong, P.M.; Anderson, J.F.; Vossbrinck, C.R. Host Feeding Patterns of Culex Mosquitoes and West Nile Virus Transmission, Northeastern United States. Emerg. Infect. Dis. 2006, 12, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Monath, T.P. The Arboviruses: Epidemiology and Ecology. Available online: https://www.routledge.com/The-Arboviruses-Epidemiology-and-Ecology/Monath/p/book/9780367235376 (accessed on 26 December 2022).
- Brinkmann, A.; Nitsche, A.; Kohl, C. Viral Metagenomics on Blood-Feeding Arthropods as a Tool for Human Disease Surveillance. Int. J. Mol. Sci. 2016, 17, 1743. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, J.H.-O.; Shi, M.; Eden, J.-S.; Holmes, E.C.; Hesson, J.C. Meta-Transcriptomic Comparison of the RNA Viromes of the Mosquito Vectors Culex Pipiens and Culex Torrentium in Northern Europe. Viruses 2019, 11, 1033. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Beller, L.; Deboutte, W.; Yinda, K.C.; Delang, L.; Vega-Rúa, A.; Failloux, A.-B.; Matthijnssens, J. Stable Distinct Core Eukaryotic Viromes in Different Mosquito Species from Guadeloupe, Using Single Mosquito Viral Metagenomics. Microbiome 2019, 7, 121. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, M.; Altan, E.; Deng, X.; Barker, C.M.; Fang, Y.; Coffey, L.L.; Delwart, E. Virome of > 12 Thousand Culex Mosquitoes from throughout California. Virology 2018, 523, 74–88. [Google Scholar] [CrossRef]
- Feng, G.; Zhang, J.; Zhang, Y.; Li, C.; Zhang, D.; Li, Y.; Zhou, H.; Li, N.; Xiao, P. Metagenomic Analysis of Togaviridae in Mosquito Viromes Isolated from Yunnan Province in China Reveals Genes from Chikungunya and Ross River Viruses. Front. Cell Infect. Microbiol. 2022, 12, 849662. [Google Scholar] [CrossRef]
- Weaver, S.C.; Reisen, W.K. Present and Future Arboviral Threats. Antivir. Res. 2010, 85, 328–345. [Google Scholar] [CrossRef]
- Kwok, K.T.T.; Nieuwenhuijse, D.F.; Phan, M.V.T.; Koopmans, M.P.G. Virus Metagenomics in Farm Animals: A Systematic Review. Viruses 2020, 12, 107. [Google Scholar] [CrossRef]
- Baker, R.E.; Mahmud, A.S.; Miller, I.F.; Rajeev, M.; Rasambainarivo, F.; Rice, B.L.; Takahashi, S.; Tatem, A.J.; Wagner, C.E.; Wang, L.-F.; et al. Infectious Disease in an Era of Global Change. Nat. Rev. Microbiol. 2022, 20, 193–205. [Google Scholar] [CrossRef]
- Rasambainarivo, F.; Goodman, S.M. Disease Risk to Endemic Animals from Introduced Species on Madagascar. In Fowler’s Zoo and Wild Animal Medicine Current Therapy; Saunders: Philadelphia, PA, USA, 2019; Volume 9, pp. 292–297. [Google Scholar] [CrossRef]
- Myers, N.; Mittermeier, R.A.; Mittermeier, C.G.; da Fonseca, G.A.B.; Kent, J. Biodiversity Hotspots for Conservation Priorities. Nature 2000, 403, 853–858. [Google Scholar] [CrossRef]
- Tantely, M.L.; Le Goff, G.; Boyer, S.; Fontenille, D. An Updated Checklist of Mosquito Species (Diptera: Culicidae) from Madagascar. Parasite 2016, 23, 20. [Google Scholar] [CrossRef]
- Fontenille, D.; Mathiot, C.; Rodhain, F.; Coulanges, P. Arbovirus infections on the island of Nosy-Be; serologic and entomologic findings. Arch. Inst. Pasteur Madag. 1988, 54, 101–115. [Google Scholar]
- Fontenille, D. Arbovirus transmission cycles in Madagascar. Arch. Inst. Pasteur Madag. 1989, 55, 1–317. [Google Scholar]
- Jeanmaire, E.M.; Rabenarivahiny, R.; Biarmann, M.; Rabibisoa, L.; Ravaomanana, F.; Randriamparany, T.; Andriamandimby, S.F.; Diaw, C.S.; Fenozara, P.; de La Rocque, S.; et al. Prevalence of Rift Valley Fever Infection in Ruminants in Madagascar after the 2008 Outbreak. Vector Borne Zoonotic Dis. 2011, 11, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Andriamandimby, S.F.; Viarouge, C.; Ravalohery, J.-P.; Reynes, J.-M.; Sailleau, C.; Tantely, M.L.; Elissa, N.; Cardinale, E.; Sall, A.A.; Zientara, S.; et al. Detection in and Circulation of Bluetongue Virus among Domestic Ruminants in Madagascar. Vet. Microbiol. 2015, 176, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Ratovonjato, J.; Olive, M.-M.; Tantely, L.M.; Andrianaivolambo, L.; Tata, E.; Razainirina, J.; Jeanmaire, E.; Reynes, J.-M.; Elissa, N. Detection, Isolation, and Genetic Characterization of Rift Valley Fever Virus from Anopheles (Anopheles) Coustani, Anopheles (Anopheles) Squamosus, and Culex (Culex) Antennatus of the Haute Matsiatra Region, Madagascar. Vector Borne Zoonotic Dis. 2011, 11, 753–759. [Google Scholar] [CrossRef]
- Chevalier, V.; Marsot, M.; Molia, S.; Rasamoelina, H.; Rakotondravao, R.; Pedrono, M.; Lowenski, S.; Durand, B.; Lecollinet, S.; Beck, C. Serological Evidence of West Nile and Usutu Viruses Circulation in Domestic and Wild Birds in Wetlands of Mali and Madagascar in 2008. Int. J. Environ. Res. Public Health 2020, 17, 1998. [Google Scholar] [CrossRef]
- Institut Pasteur de Madagascar. Rapport Annuel 2021, Rapport Annuel de l’Institut Pasteur de Madagascar. 2022. Available online: https://www.pasteur.mg/wp-content/uploads/2022/09/Rapport-dactivites-IPM_2021.pdf (accessed on 5 July 2023).
- Pérot, P.; Bigot, T.; Temmam, S.; Regnault, B.; Eloit, M. Microseek: A Protein-Based Metagenomic Pipeline for Virus Diagnostic and Discovery. Viruses 2022, 14, 1990. [Google Scholar] [CrossRef]
- Bigot, T.; Temmam, S.; Pérot, P.; Eloit, M. RVDB-Prot, a Reference Viral Protein Database and Its HMM Profiles. F1000Research 2019, 8, 530. [Google Scholar] [CrossRef]
- Goodacre, N.; Aljanahi, A.; Nandakumar, S.; Mikailov, M.; Khan, A.S. A Reference Viral Database (RVDB) To Enhance Bioinformatics Analysis of High-Throughput Sequencing for Novel Virus Detection. mSphere 2018, 3, e00069-18. [Google Scholar] [CrossRef]
- Oksanen, J.; Simpson, G.L.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Solymos, P.; Stevens, M.H.H.; Szoecs, E.; et al. Vegan: Community Ecology Package; ResearchGate: Berlin, Germany, 2022. [Google Scholar]
- Krebs, C.J. Ecological Methodology; Harpercollins College Div.: New York City, NY, USA, 1989; ISBN 978-0-06-043784-8. [Google Scholar]
- Volant, S.; Lechat, P.; Woringer, P.; Motreff, L.; Campagne, P.; Malabat, C.; Kennedy, S.; Ghozlane, A. SHAMAN: A User-Friendly Website for Metataxonomic Analysis from Raw Reads to Statistical Analysis. BMC Bioinform. 2020, 21, 345. [Google Scholar] [CrossRef] [PubMed]
- Stephanie, G. Benjamini-Hochberg Procedure. Available online: https://www.statisticshowto.com/benjamini-hochberg-procedure/ (accessed on 5 July 2023).
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Sólymos, P.; Stevens, M.H.H.; Wagner, H. Vegan: Community Ecology Package. 2012. Available online: http://CRAN.R-project.org/package=vegan (accessed on 5 July 2023).
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (ITOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, K.; Isawa, H.; Tsuda, Y.; Yano, K.; Sasaki, T.; Yuda, M.; Takasaki, T.; Kobayashi, M.; Sawabe, K. Genetic Characterization of a New Insect Flavivirus Isolated from Culex Pipiens Mosquito in Japan. Virology 2007, 359, 405–414. [Google Scholar] [CrossRef]
- Cholleti, H.; Hayer, J.; Abilio, A.P.; Mulandane, F.C.; Verner-Carlsson, J.; Falk, K.I.; Fafetine, J.M.; Berg, M.; Blomström, A.-L. Discovery of Novel Viruses in Mosquitoes from the Zambezi Valley of Mozambique. PLoS ONE 2016, 11, e0162751. [Google Scholar] [CrossRef]
- Xia, H.; Wang, Y.; Shi, C.; Atoni, E.; Zhao, L.; Yuan, Z. Comparative Metagenomic Profiling of Viromes Associated with Four Common Mosquito Species in China. Virol. Sin. 2018, 33, 59. [Google Scholar] [CrossRef]
- Frey, K.G.; Biser, T.; Hamilton, T.; Santos, C.J.; Pimentel, G.; Mokashi, V.P.; Bishop-Lilly, K.A. Bioinformatic Characterization of Mosquito Viromes within the Eastern United States and Puerto Rico: Discovery of Novel Viruses. Evol. Bioinform. 2016, 12, 1. [Google Scholar] [CrossRef]
- Coffey, L.L.; Page, B.L.; Greninger, A.L.; Herring, B.L.; Russell, R.C.; Doggett, S.L.; Haniotis, J.; Wang, C.; Deng, X.; Delwart, E.L. Enhanced Arbovirus Surveillance with Deep Sequencing: Identification of Novel Rhabdoviruses and Bunyaviruses in Australian Mosquitoes. Virology 2014, 448, 146–158. [Google Scholar] [CrossRef]
- Li, C.-X.; Shi, M.; Tian, J.-H.; Lin, X.-D.; Kang, Y.-J.; Chen, L.-J.; Qin, X.-C.; Xu, J.; Holmes, E.C.; Zhang, Y.-Z. Unprecedented Genomic Diversity of RNA Viruses in Arthropods Reveals the Ancestry of Negative-Sense RNA Viruses. eLife 2015, 4, e05378. [Google Scholar] [CrossRef]
- Shi, M.; Neville, P.; Nicholson, J.; Eden, J.-S.; Imrie, A.; Holmes, E.C. High-Resolution Metatranscriptomics Reveals the Ecological Dynamics of Mosquito-Associated RNA Viruses in Western Australia. J. Virol. 2017, 91, e00680-17. [Google Scholar] [CrossRef]
- Coatsworth, H.; Bozic, J.; Carrillo, J.; Buckner, E.A.; Rivers, A.R.; Dinglasan, R.R.; Mathias, D.K. Intrinsic Variation in the Vertically Transmitted Core Virome of the Mosquito Aedes Aegypti. Mol. Ecol. 2022, 31, 2545–2561. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, K.; Dovrolis, N.; Kouvela, A.; Kassela, K.; Rosa Freitas, M.G.; Nearchou, A.; de Courcy Williams, M.; Veletza, S.; Karakasiliotis, I. Defining Virus-Carrier Networks That Shape the Composition of the Mosquito Core Virome of a Local Ecosystem. Virus Evol. 2022, 8, veac036. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Districts | Villages Names | Description of Landscape |
|---|---|---|
| Mahajanga I | Urban Commune of Mahajanga 15′43′10″80″ S 46′18′20″25″ E | 35 m above sea level (asl); the historical neighborhood of the city, with large modern cement houses/building made of cement occupying almost 100% of the area. |
| Mahajanga I | Antanimalandy 15′42′53″22″ S 46′21′32″97″ E | 17 m asl; made up of cement houses, wooden houses, and sheet metal houses, which occupy almost 50% of the area. |
| Mahajanga II | Belobaka 15′42′12″21″ S 46′23′34″24″ E | 15 m asl; the majority of habitations are made of sheet metal house and occupy 30% of the area. A lot of mango trees, swamp, and rice fields are observed. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bennouna, A.; Tantely, M.L.; Raharinosy, V.; Andriamandimby, S.F.; Bigot, T.; Chrétien, D.; Jacquemet, E.; Volant, S.; Temmam, S.; Dussart, P.; et al. Comprehensive Characterization of Viral Diversity of Female Mosquitoes in Madagascar. Viruses 2023, 15, 1852. https://doi.org/10.3390/v15091852
Bennouna A, Tantely ML, Raharinosy V, Andriamandimby SF, Bigot T, Chrétien D, Jacquemet E, Volant S, Temmam S, Dussart P, et al. Comprehensive Characterization of Viral Diversity of Female Mosquitoes in Madagascar. Viruses. 2023; 15(9):1852. https://doi.org/10.3390/v15091852
Chicago/Turabian StyleBennouna, Amal, Michael Luciano Tantely, Vololoniaina Raharinosy, Soa Fy Andriamandimby, Thomas Bigot, Delphine Chrétien, Elise Jacquemet, Stevenn Volant, Sarah Temmam, Philippe Dussart, and et al. 2023. "Comprehensive Characterization of Viral Diversity of Female Mosquitoes in Madagascar" Viruses 15, no. 9: 1852. https://doi.org/10.3390/v15091852
APA StyleBennouna, A., Tantely, M. L., Raharinosy, V., Andriamandimby, S. F., Bigot, T., Chrétien, D., Jacquemet, E., Volant, S., Temmam, S., Dussart, P., Lacoste, V., Girod, R., & Eloit, M. (2023). Comprehensive Characterization of Viral Diversity of Female Mosquitoes in Madagascar. Viruses, 15(9), 1852. https://doi.org/10.3390/v15091852