
Porcine Reproductive and Respiratory Syndrome (PRRSV2) Viral Diversity within a Farrow-to-Wean Farm Cohort Study

 , ,
, ,  ,
,  ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
3. Results
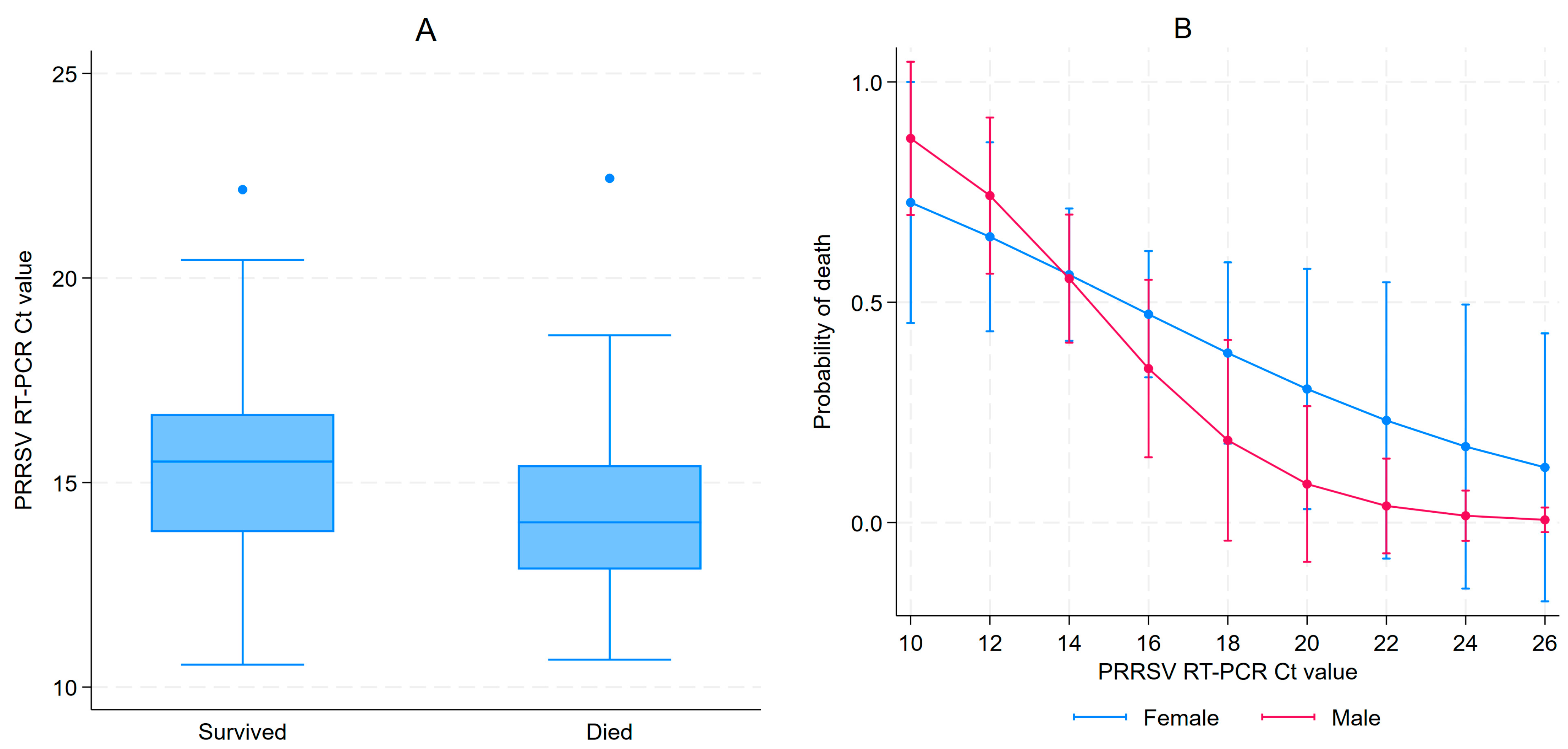
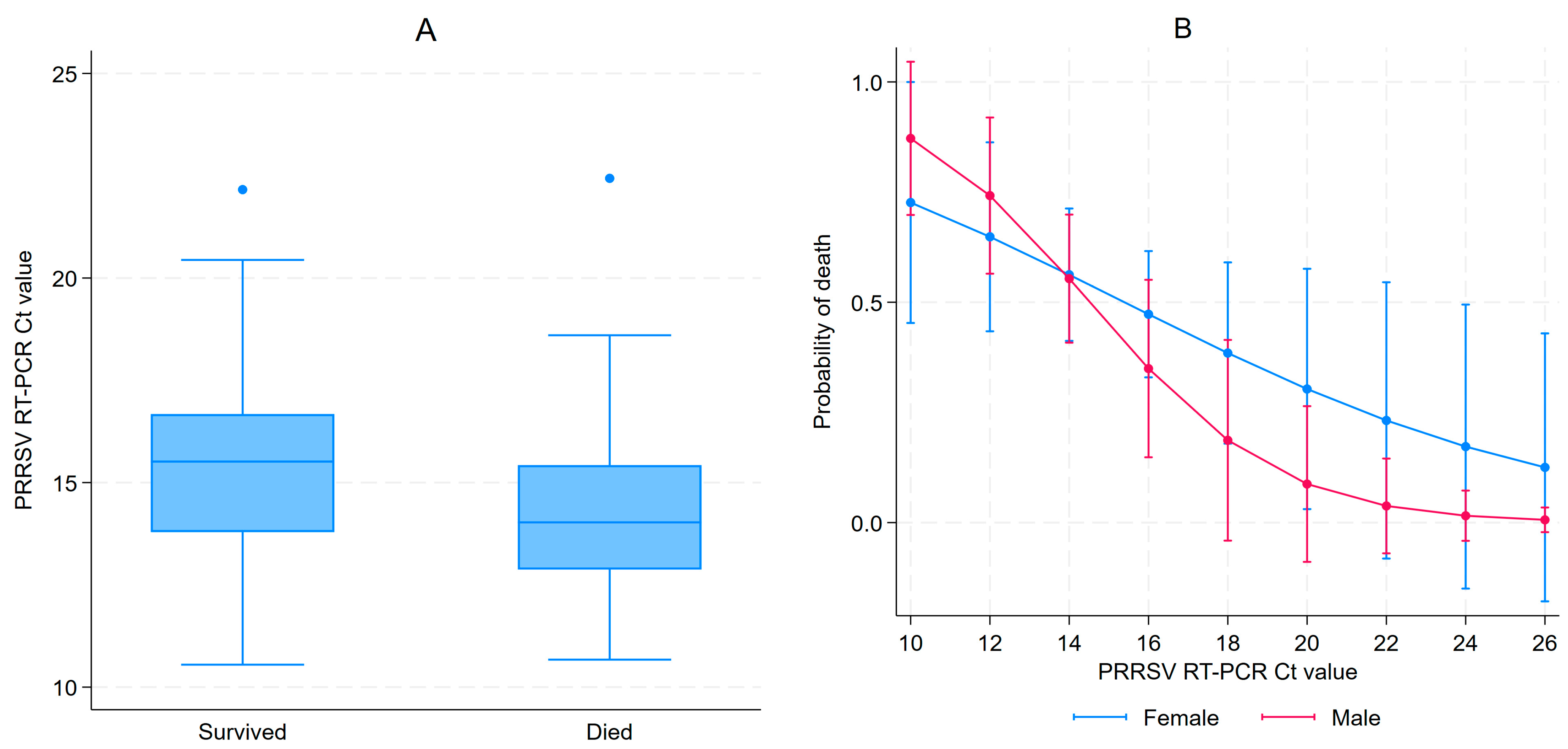
3.1. PRRSV RT-PCR Results
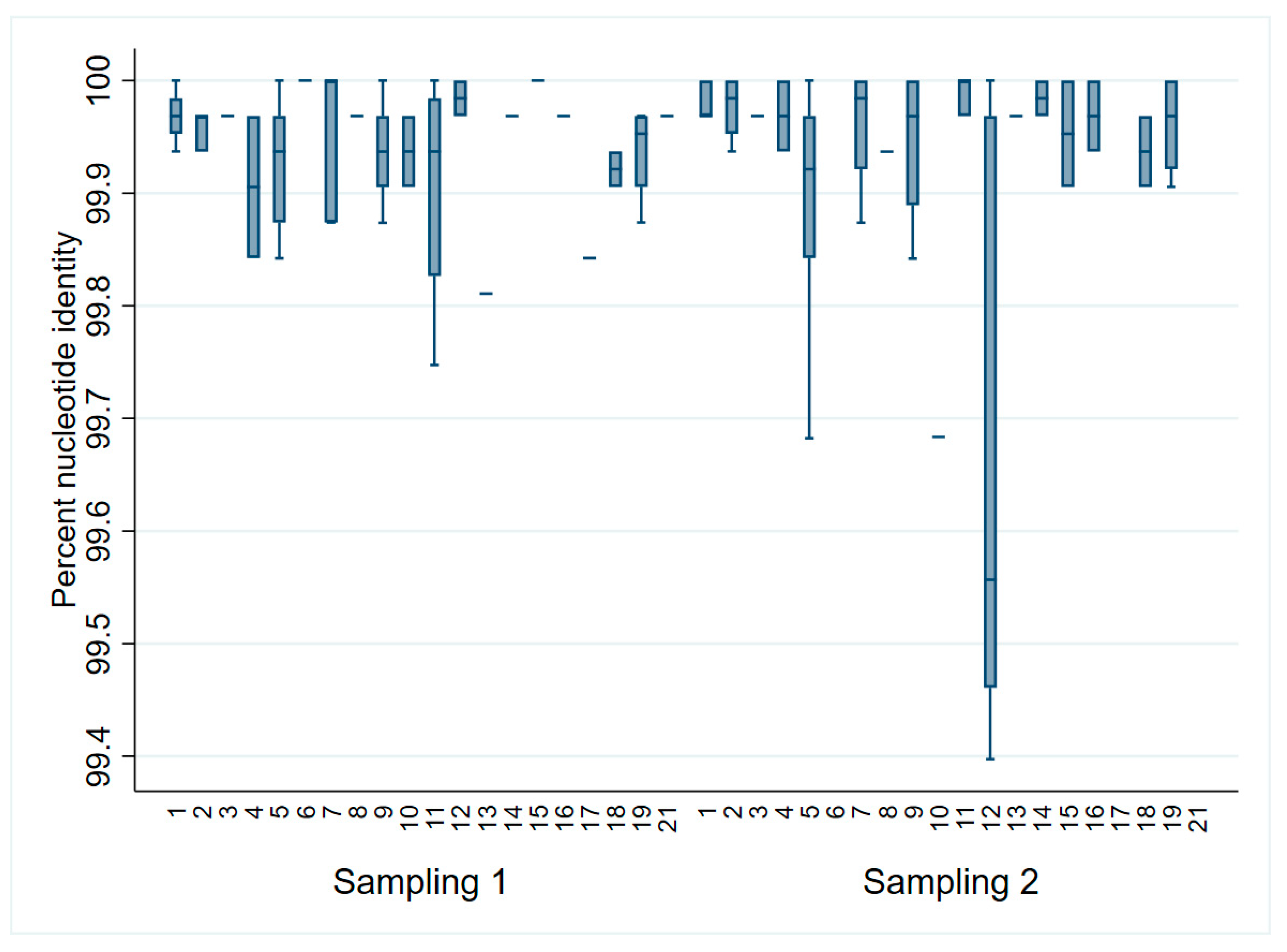
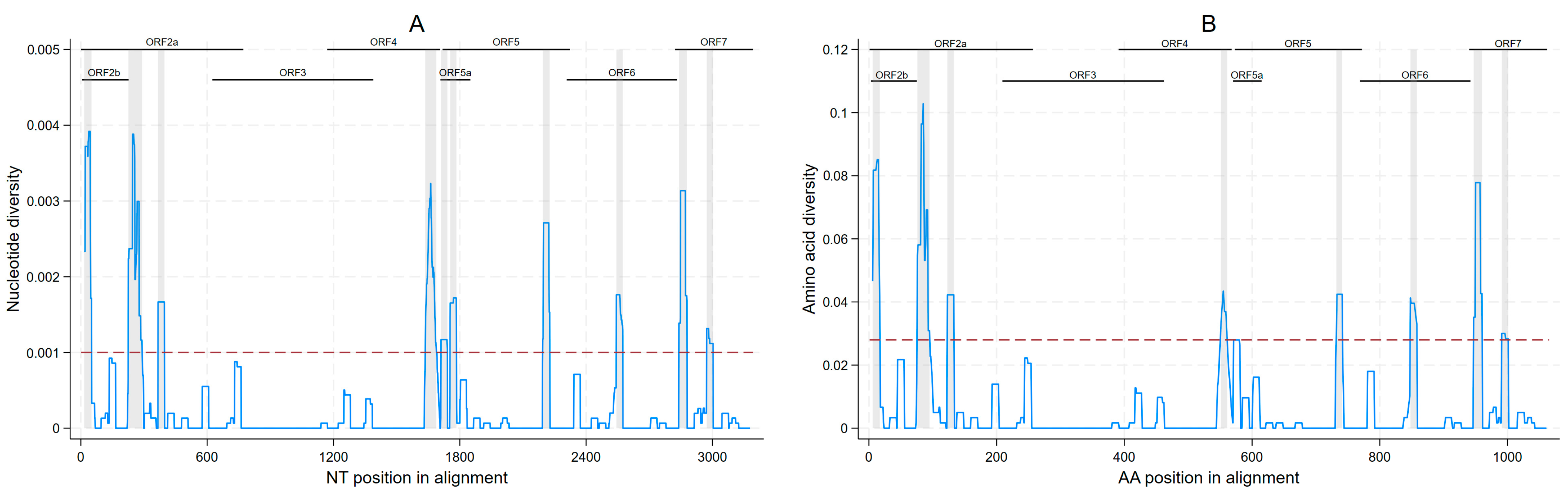
3.2. ONT Sequencing Results
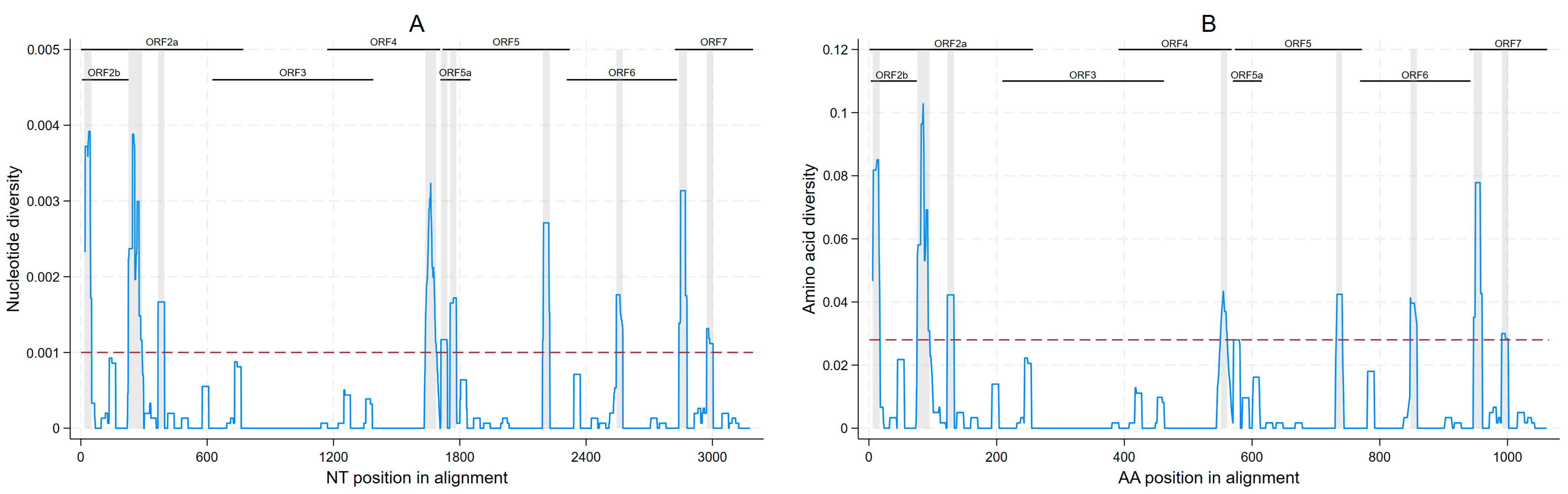
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Keffaber, K. Reproductive failure of unknown etiology. Am. Assoc. Swine Pract. News 1989, a, 1–9. [Google Scholar]
- Benfield, D.; Nelson, E.; Collins, J.; Harris, L.; Goyal, S.; Robison, D.; Christianson, W.; Morrison, R.; Gorcyca, D.; Chladek, D. Characterization of swine infertility and respiratory syndrome (SIRS) virus (isolate ATCC VR-2332). J. Vet. Diagn. Invest. 1992, 4, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Kikuti, M.; Sanhueza, J.; Vilalta, C.; Paploski, I.A.D.; VanderWaal, K.; Corzo, C.A. Porcine reproductive and respiratory syndrome virus 2 (PRRSV-2) genetic diversity and occurrence of wild type and vaccine-like strains in the United States swine industry. PLoS ONE 2021, 16, e0259531. [Google Scholar] [CrossRef] [PubMed]
- Cortey, M.; Díaz, I.; Martín-Valls, G.; Mateu, E. Next-generation sequencing as a tool for the study of Porcine reproductive and respiratory syndrome virus (PRRSV) macro- and micro- molecular epidemiology. Vet. Microbiol. 2017, 209, 5–12. [Google Scholar] [CrossRef]
- Wesley, R.; Mengeling, W.; Lager, K.; Clouser, D.; Landgraf, J.; Frey, M. Differentiation of a porcine reproductive and respiratory syndrome virus vaccine strain from North American field strains by restriction fragment length polymorphism analysis of ORF 5. J. Vet. Diagn. Invest. 1998, 10, 140–144. [Google Scholar] [CrossRef]
- Chang, C.; Yoon, K.; Zimmerman, J.; Harmon, K.; Dixon, P.; Dvorak, C.; Murtaugh, M. Evolution of Porcine Reproductive and Respiratory Syndrome Virus during Sequential Passages in Pigs. J. Virol. 2002, 76, 4750–4763. [Google Scholar] [CrossRef]
- Murtaugh, M.; Stadejek, T.; Abrahante, J.; Lam, T.; Leung, F. The ever-expanding diversity of porcine reproductive and respiratory syndrome virus. Virus Res. 2010, 154, 18–30. [Google Scholar] [CrossRef]
- Shi, M.; Lam, T.T.-Y.; Hon, C.-C.; Murtaugh, M.P.; Davies, P.R.; Hui, R.K.-H.; Li, J.; Wong, L.T.-W.; Yip, C.-W.; Jiang, J.-W.; et al. Phylogeny-Based Evolutionary, Demographical, and Geographical Dissection of North American Type 2 Porcine Reproductive and Respiratory Syndrome Viruses. J. Virol. 2010, 84, 8700–8711. [Google Scholar] [CrossRef]
- Paploski, I.A.D.; Corzo, C.; Rovira, A.; Murtaugh, M.P.; Sanhueza, J.M.; Vilalta, C.; Schroeder, D.C.; VanderWaal, K. Temporal Dynamics of Co-circulating Lineages of Porcine Reproductive and Respiratory Syndrome Virus. Front. Microbiol. 2019, 10, 2486. [Google Scholar] [CrossRef]
- Paploski, I.A.D.; Pamornchainavakul, N.; Makau, D.N.; Rovira, A.; Corzo, C.A.; SchroederSchroeder, D.C.; Cheeran, M.C.-J.; Doeschl-Wilson, A.; Kao, R.R.; Lycett, S.; et al. Phylogenetic Structure and Sequential Dominance of Sub-Lineages of PRRSV Type-2 Lineage 1 in the United States. Vaccines 2021, 9, 608. [Google Scholar] [CrossRef]
- Cha, S.; Chang, C.; Yoon, K. Instability of the Restriction Fragment Length Polymorphism Pattern of Open Reading Frame 5 of Porcine Reproductive and Respiratory Syndrome Virus during Sequential Pig-to-Pig Passages. J. Clin. Microbiol. 2004, 42, 4462–4467. [Google Scholar] [CrossRef] [PubMed]
- Kwon, T.; Yoo, S.; Lee, D.; Sunwoo, S.; Je, S.; Park, J.; Kim, M.; Park, C.; Lyoo, Y. Differential evolution of antigenic regions of porcine reproductive and respiratory syndrome virus 1 before and after vaccine introduction. Virus Res. 2018, 260, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Brar, M.; Mang, S.; Hui, R.-H.; Leung, F.-C. Genomic Evolution of Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) Isolates Revealed by Deep Sequencing. PLoS ONE 2014, 9, e88807. [Google Scholar] [CrossRef]
- Domingo, E.; Schuster, P. What Is a Quasispecies? Historical Origins and Current Scope; Springer Nature: Berlin/Heidelberg, Germany, 2015; pp. 1–22. [Google Scholar]
- Hanada, K.; Suzuki, Y.; Nakane, T.; Hirose, O.; Gojobori, T. The origin and evolution of porcine reproductive and respiratory syndrome viruses. Mol. Biol. Evol. 2005, 22, 1024–1031. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, T.; Lowe, J.; Milburn, S.; Firkins, L. Quasispecies variation of porcine reproductive and respiratory syndrome virus during natural infection. Virology 2003, 317, 197–207. [Google Scholar] [CrossRef]
- Holtkamp, D.J.; Polson, D.D.; Torremorell, M.; Morrison, B.; Classen, D.M.; Becton, L.; Henry, S.; Rodibaugh, M.T.; Rowland, R.R.; Snelson, H.; et al. Terminology for classifying the porcine reproductive and respiratory syndrome virus (PRRSV) status of swine herds. Tierarztl. Prax. Ausg. G. Grosstiere. Nutztiere. 2011, 39, 101–112. [Google Scholar]
- Pileri, E.; Mateu, E. Review on the transmission porcine reproductive and respiratory syndrome virus between pigs and farms and impact on vaccination. Vet. Res. 2016, 47, 108. [Google Scholar] [CrossRef]
- Rowland, R.; Lawson, S.; Rossow, K.; Benfield, D. Lymphoid tissue tropism of porcine reproductive and respiratory syndrome virus replication during persistent infection of pigs originally exposed to virus in utero. Vet. Microbiol. 2003, 96, 219–235. [Google Scholar] [CrossRef]
- Dee, S.; Joo, H. Strategies to control PRRS: A summary of field and research experiences. Vet. Microbiol. 1997, 55, 347–353. [Google Scholar] [CrossRef]
- Nielsen, J.; Botner, A.; Bille-Hansen, V.; Oleksiewicz, M.; Storgaard, T. Experimental inoculation of late term pregnant sows with a field isolate of porcine reproductive and respiratory syndrome vaccine-derived virus. Vet. Microbiol. 2002, 84, 1–13. [Google Scholar] [CrossRef]
- Schroeder, D.C.; Odogwu, N.M.; Kevill, J.; Yang, M.; Krishna, V.D.; Kikuti, M.; Pamornchainavakul, N.; Vilalta, C.; Sanhueza, J.; Corzo, C.A.; et al. Phylogenetically Distinct Near-Complete Genome Sequences of Porcine Reproductive and Respiratory Syndrome Virus Type 2 Variants from Four Distinct Disease Outbreaks at U.S. Swine Farms over the Past 6 Years. Microbiol. Resour. Announc. 2021, 10, e0026021. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Leng, C.; Ding, Y.; Zhai, H.; Li, Z.; Xiang, L.; Zhang, W.; Liu, C.; Li, M.; Chen, J.; et al. Characterization of newly emerged NADC30-like strains of porcine reproductive and respiratory syndrome virus in China. Arch. Virol. 2019, 164, 401–411. [Google Scholar] [CrossRef]
- Nei, M.; Li, W.H. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. USA 1979, 76, 5269–5273. [Google Scholar] [CrossRef] [PubMed]
- Morais, I.J.; Polveiro, R.C.; Souza, G.M.; Bortolin, D.I.; Sassaki, F.T.; Lima, A.T.M. The global population of SARS-CoV-2 is composed of six major subtypes. Sci. Rep. 2020, 10, 18289. [Google Scholar] [CrossRef]
- StataCorp. Stata Statistical Software: Release 15; StataCorp LLC: College Station, TX, USA, 2017. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Vilalta, C.; Sanhueza, J.; Alvarez, J.; Murray, D.; Torremorell, M.; Corzo, C.; Morrison, R. Use of processing fluids and serum samples to characterize porcine reproductive and respiratory syndrome virus dynamics in 3 day-old pigs. Vet. Microbiol. 2018, 225, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Klinge, K.L.; Vaughn, E.M.; Roof, M.B.; Bautista, E.M.; Murtaugh, M.P. Age-dependent resistance to Porcine reproductive and respiratory syndrome virus replication in swine. Virol. J. 2009, 6, 177. [Google Scholar] [CrossRef]
- Clilverd, H.; Cortey, M.; Martín-Valls, G.; Mateu, E. Characterization of PRRSV1 transmission routes in an endemic farm identifies conserved phylogenetic clusters. In Proceedings of the North American PRRS Symposium, Manhattan, KS, USA, 11 December 2019; p. 66. [Google Scholar]
- Pedersen, C.-E.T.; Frandsen, P.; Wekesa, S.N.; Heller, R.; Sangula, A.K.; Wadsworth, J.; Knowles, N.J.; Muwanika, V.B.; Siegismund, H.R. Time Clustered Sampling Can Inflate the Inferred Substitution Rate in Foot-And-Mouth Disease Virus Analyses. PLoS ONE 2015, 10, e0143605. [Google Scholar] [CrossRef]
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef]
- Zhou, L.; Yang, Y.; Xia, Q.; Guan, Z.; Zhang, J.; Li, B.; Qiu, Y.; Liu, K.; Shao, D.; Ma, Z.; et al. Genetic characterization of porcine reproductive and respiratory syndrome virus from Eastern China during 2017–2022. Front. Microbiol. 2022, 13, 971817. [Google Scholar] [CrossRef]
- Delisle, B.; Gagnon, C.A.; Lambert, M.-È.; D’Allaire, S. Porcine reproductive and respiratory syndrome virus diversity of Eastern Canada swine herds in a large sequence dataset reveals two hypervariable regions under positive selection. Infect. Genet. Evol. 2012, 12, 1111–1119. [Google Scholar] [CrossRef]
- Forsberg, R. Divergence time of porcine reproductive and respiratory syndrome virus subtypes. Mol. Biol. Evol. 2005, 22, 2131–2134. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.-Y.; Campler, M.R.; Schroeder, D.C.; Yang, M.; Mor, S.K.; Ferreira, J.B.; Arruda, A.G. Detection of Multiple Lineages of PRRSV in Breeding and Growing Swine Farms. Front. Vet. Sci. 2022, 9, 884733. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Ge, X.; Zhang, Y.; Guo, X.; Han, J.; Zhou, L.; Yang, H. Genetic Characteristics of Three Single-Farm-Isolated Porcine Reproductive and Respiratory Syndrome Viruses with Novel Recombination among NADC30-Like, JXA1-Like, and QYYZ-Like Strains. Transbound. Emerg. Dis. 2023, 2023, 8871321. [Google Scholar] [CrossRef]
- Clilverd, H.; Martín-Valls, G.; Li, Y.; Martín, M.; Cortey, M.; Mateu, E. Infection dynamics, transmission, and evolution after an outbreak of porcine reproductive and respiratory syndrome virus. Front. Microbiol. 2023, 14, 1109881. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
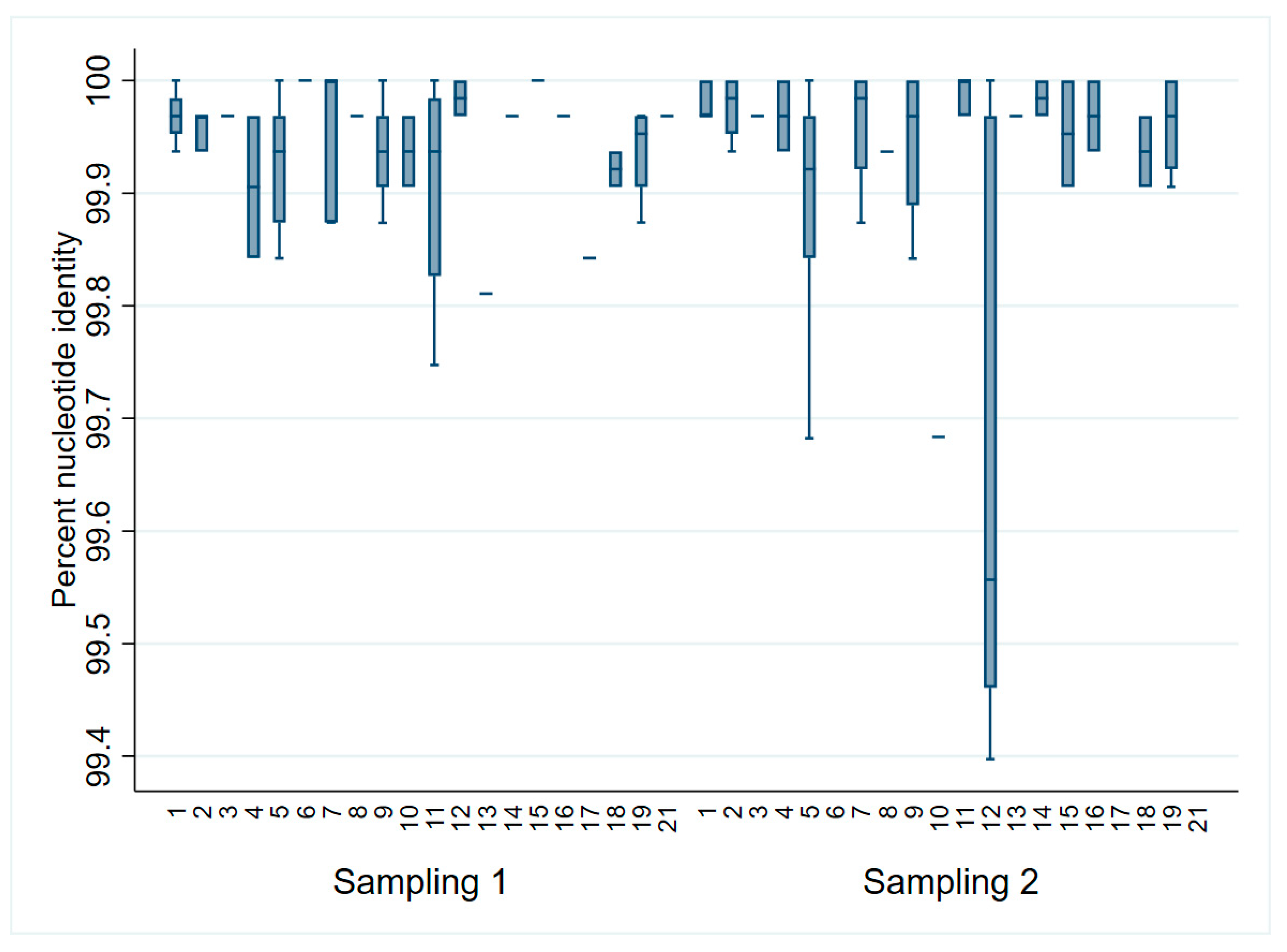
| Percent Identity | Sampling 1 (S1) | Sampling 2 (S2) | Within Animals (S1 × S2) | |||
|---|---|---|---|---|---|---|
| Overall | Within Litter | Overall | Within Litter | |||
| ORF2–7 | Minimum | 99.84 | 99.88 | 99.57 | 99.57 | 99.65 |
| Maximum | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | |
| ORF2a | Minimum | 99.21 | 99.34 | 98.94 | 99.07 | 99.21 |
| Maximum | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | |
| ORF2b | Minimum | 99.07 | 99.07 | 98.60 | 99.06 | 99.06 |
| Maximum | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | |
| ORF3 | Minimum | 99.74 | 99.87 | 99.74 | 99.74 | 99.87 |
| Maximum | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | |
| ORF4 | Minimum | 99.63 | 99.81 | 95.81 | 95.81 | 97.26 |
| Maximum | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | |
| ORF5a | Minimum | 97.74 | 98.53 | 98.53 | 98.53 | 98.53 |
| Maximum | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | |
| ORF5 | Minimum | 99.50 | 99.50 | 99.66 | 99.67 | 99.66 |
| Maximum | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | |
| ORF6 | Minimum | 99.62 | 99.81 | 99.61 | 99.80 | 99.80 |
| Maximum | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | |
| ORF7 | Minimum | 99.46 | 99.73 | 99.73 | 99.73 | 100.00 |
| Maximum | 100.00 | 100.00 | 100.00 | 100.00 | 100.00 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kikuti, M.; Vilalta, C.; Sanhueza, J.; Pamornchainavakul, N.; Kevill, J.; Yang, M.; Paploski, I.A.D.; Lenskaia, T.; Odogwu, N.M.; Kiehne, R.; et al. Porcine Reproductive and Respiratory Syndrome (PRRSV2) Viral Diversity within a Farrow-to-Wean Farm Cohort Study. Viruses 2023, 15, 1837. https://doi.org/10.3390/v15091837
Kikuti M, Vilalta C, Sanhueza J, Pamornchainavakul N, Kevill J, Yang M, Paploski IAD, Lenskaia T, Odogwu NM, Kiehne R, et al. Porcine Reproductive and Respiratory Syndrome (PRRSV2) Viral Diversity within a Farrow-to-Wean Farm Cohort Study. Viruses. 2023; 15(9):1837. https://doi.org/10.3390/v15091837
Chicago/Turabian StyleKikuti, Mariana, Carles Vilalta, Juan Sanhueza, Nakarin Pamornchainavakul, Jessica Kevill, My Yang, Igor A. D. Paploski, Tatiana Lenskaia, Nkechi M. Odogwu, Ross Kiehne, and et al. 2023. "Porcine Reproductive and Respiratory Syndrome (PRRSV2) Viral Diversity within a Farrow-to-Wean Farm Cohort Study" Viruses 15, no. 9: 1837. https://doi.org/10.3390/v15091837
APA StyleKikuti, M., Vilalta, C., Sanhueza, J., Pamornchainavakul, N., Kevill, J., Yang, M., Paploski, I. A. D., Lenskaia, T., Odogwu, N. M., Kiehne, R., VanderWaal, K., Schroeder, D., & Corzo, C. A. (2023). Porcine Reproductive and Respiratory Syndrome (PRRSV2) Viral Diversity within a Farrow-to-Wean Farm Cohort Study. Viruses, 15(9), 1837. https://doi.org/10.3390/v15091837