

Recurring Trans-Atlantic Incursion of Clade 2.3.4.4b H5N1 Viruses by Long Distance Migratory Birds from Northern Europe to Canada in 2022/2023
 ,
,  , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Samples
2.2. Nanopore Sequencing and Genome Assembly
2.3. Phylogenetic Analysis
3. Results and Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hill, N.J.; Bishop, M.A.; Trovão, N.S.; Ineson, K.M.; Schaefer, A.L.; Puryear, W.B.; Zhou, K.; Foss, A.D.; Clark, D.E.; MacKenzie, K.G.; et al. Ecological Divergence of Wild Birds Drives Avian Influenza Spillover and Global Spread. PLoS Pathog. 2022, 18, e1010062. [Google Scholar] [CrossRef]
- Xu, X.; Subbarao, K.; Cox, N.J.; Guo, Y. Genetic Characterization of the Pathogenic Influenza A/Goose/Guangdong/1/96 (H5N1) Virus: Similarity of its Hemagglutinin Gene to those of H5N1 Viruses from the 1997 Outbreaks in Hong Kong. Virology 1999, 261, 15–19. [Google Scholar] [CrossRef]
- Chen, H.; Smith, G.J.; Li, K.S.; Wang, J.; Fan, X.H.; Rayner, J.M.; Vijaykrishna, D.; Zhang, J.X.; Zhang, L.J.; Guo, C.T.; et al. Establishment of Multiple Sublineages of H5N1 Influenza Virus in Asia: Implications for Pandemic Control. Proc. Natl. Acad. Sci. USA 2006, 103, 2845–2850. [Google Scholar] [CrossRef]
- Lin, R.; Lu, L.; Lycett, S.; Liu, W.; Li, J. Dealing with Highly Pathogenic Avian Influenza: An Impending Crisis. Innovation 2021, 2, 100084. [Google Scholar] [CrossRef] [PubMed]
- Pohlmann, A.; King, J.; Fusaro, A.; Zecchin, B.; Banyard, A.C.; Brown, I.H.; Byrne, A.M.P.; Beerens, N.; Liang, Y.; Heutink, R.; et al. Has Epizootic Become Enzootic? Evidence for a Fundamental Change in the Infection Dynamics of Highly Pathogenic Avian Influenza in Europe, 2021. mBio 2022, 13, e00609–e00622. [Google Scholar] [CrossRef]
- Beerens, N.; Koch, G.; Heutink, R.; Harders, F.; Vries, D.P.E.; Ho, C.; Bossers, A.; Elbers, A. Novel Highly Pathogenic Avian Influenza A(H5N6) Virus in the Netherlands, December 2017. Emerg. Infect. Dis. 2018, 24, 770–773. [Google Scholar] [CrossRef]
- Śmietanka, K.; Świętoń, E.; Kozak, E.; Wyrostek, K.; Tarasiuk, K.; Tomczyk, G.; Konopka, B.; Welz, M.; Domańska-Blicharz, K.; Niemczuk, K.; et al. Highly Pathogenic Avian Influenza H5N8 in Poland in 2019-2020. J. Vet. Res. 2020, 64, 469–476. [Google Scholar] [CrossRef]
- Bevins, S.N.; Shriner, S.A.; Cumbee, J.C., Jr.; Dilione, K.E.; Douglass, K.E.; Ellis, J.W.; Killian, M.L.; Torchetti, M.K.; Lenoch, J.B. Intercontinental Movement of Highly Pathogenic Avian Influenza A(H5N1) Clade 2.3.4.4 Virus to the United States, 2021. Emerg. Infect. Dis. 2022, 28, 1006–1011. [Google Scholar] [CrossRef]
- Caliendo, V.; Lewis, N.S.; Pohlmann, A.; Baillie, S.R.; Banyard, A.C.; Beer, M.; Brown, I.H.; Fouchier, R.A.M.; Hansen, R.D.E.; Lameris, T.K.; et al. Transatlantic Spread of Highly Pathogenic Avian Influenza H5N1 by Wild Birds from Europe to North America in 2021. Sci. Rep. 2022, 12, 11729. [Google Scholar] [CrossRef]
- Rijks, J.M.; Leopold, M.F.; Kühn, S.; In‘t Veld, R.; Schenk, F.; Brenninkmeijer, A.; Lilipaly, S.J.; Ballmann, M.Z.; Kelder, L.; de Jong, J.W.; et al. Mass Mortality Caused by Highly Pathogenic Influenza A(H5N1) Virus in Sandwich Terns, the Netherlands, 2022. Emerg. Infect. Dis. 2022, 28, 2538–2542. [Google Scholar] [CrossRef]
- Lewis, N.S.; Banyard, A.C.; Whittard, E.; Karibayev, T.; Al Kafagi, T.; Chvala, I.; Byrne, A.; Meruyert Akberovna, S.; King, J.; Harder, T.; et al. Emergence and Spread of Novel H5N8, H5N5 and H5N1 Clade 2.3.4.4 Highly Pathogenic Avian Influenza in 2020. Emerg. Microbes Infect. 2021, 10, 148–151. [Google Scholar] [CrossRef] [PubMed]
- Günther, A.; Krone, O.; Svansson, V.; Pohlmann, A.; King, J.; Hallgrimsson, G.T.; Skarphéðinsson, K.H.; Sigurðardóttir, H.; Jónsson, S.R.; Beer, M.; et al. Iceland as Stepping Stone for Spread of Highly Pathogenic Avian Influenza Virus between Europe and North America. Emerg. Infect. Dis. 2022, 28, 2383–2388. [Google Scholar] [CrossRef]
- Verhagen, J.H.; Fouchier, R.A.M.; Lewis, N. Highly Pathogenic Avian Influenza Viruses at the Wild-Domestic Bird Interface in Europe: Future Directions for Research and Surveillance. Viruses 2021, 13, 212. [Google Scholar] [CrossRef]
- Nemeth, N.M.; Ruder, M.G.; Poulson, R.L.; Sargent, R.; Breeding, S.; Evans, M.N.; Zimmerman, J.; Hardman, R.; Cunningham, M.; Gibbs, S.; et al. Bald Eagle Mortality and Nest Failure Due to Clade 2.3.4.4 Highly Pathogenic H5N1 Influenza A Virus. Sci. Rep. 2023, 13, 191. [Google Scholar] [CrossRef] [PubMed]
- Alkie, T.N.; Lopes, S.; Hisanaga, T.; Xu, W.; Suderman, M.; Koziuk, J.; Fisher, M.; Redford, T.; Lung, O.; Joseph, T.; et al. A Threat from both Sides: Multiple Introductions of Genetically Distinct H5 HPAI Viruses into Canada via both East Asia-Australasia/Pacific and Atlantic Flyways. Virus Evol. 2022, 8, veac077. [Google Scholar] [CrossRef]
- Weingartl, H.M.; Berhane, Y.; Hisanaga, T.; Neufeld, J.; Kehler, H.; Emburry-Hyatt, C.; Hooper-McGreevy, K.; Kasloff, S.; Dalman, B.; Bystrom, J.; et al. Genetic and Pathobiologic Characterization of Pandemic H1N1 2009 Influenza Viruses from a Naturally Infected Swine herd. J. Virol. 2010, 84, 2245–2256. [Google Scholar] [CrossRef]
- Spackman, E.; Senne, D.A.; Myers, T.J.; Bulaga, L.L.; Garber, L.P.; Perdue, M.L.; Lohman, K.; Daum, L.T.; Suarez, D.L. Development of a Real-Time Reverse Transcriptase PCR Assay for Type A Influenza Virus and the Avian H5 and H7 Hemagglutinin Subtypes. J. Clin. Microbiol. 2002, 40, 3256–3260. [Google Scholar] [CrossRef]
- OIE. Avian Influenza. In Manual of Diagnostic Tests and Vaccines for Terrestrial Animals; OIE: Paris, France, 2021. [Google Scholar]
- Zhou, B.; Donnelly, M.E.; Scholes, D.T.; St George, K.; Hatta, M.; Kawaoka, Y.; Wentworth, D.E. Single-reaction Genomic Amplification Accelerates Sequencing and Vaccine Production for Classical and Swine Origin Human Influenza A Viruses. J. Virol. 2009, 83, 10309–10313. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Sagulenko, P.; Puller, V.; Neher, R.A. TreeTime: Maximum-likelihood Phylodynamic Analysis. Virus Evol. 2018, 4, vex042. [Google Scholar] [CrossRef]
- Guangchuang, Y.; David, K.S.; Huachen, Z.; Yi, G.; Tommy, T.-Y.L. GGTREE: An R Package for Visualization and Annotation of Phylogenetic Trees with Their Covariates and other Associated Data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar]
- Wang, L.G.; Lam, T.T.; Xu, S.; Dai, Z.; Zhou, L.; Feng, T.; Guo, P.; Dunn, C.W.; Jones, B.R.; Bradley, T.; et al. An R Package for Phylogenetic Tree Input and Output with Richly Annotated and Associated Data. Mol. Biol. Evol. 2020, 37, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Pasick, J.; Berhane, Y.; Joseph, T.; Bowes, V.; Hisanaga, T.; Handel, K.; Alexandersen, S. Reassortant Highly Pathogenic Influenza A H5N2 Virus Containing Gene Segments Related to Eurasian H5N8 in British Columbia, Canada, 2014. Sci. Rep. 2015, 5, 9484. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Torchetti, M.K.; Winker, K.; Ip, H.S.; Song, C.S.; Swayne, D.E. Intercontinental Spread of Asian-Origin H5N8 to North America through Beringia by Migratory Birds. J. Virol. 2015, 89, 6521–6524. [Google Scholar] [CrossRef]
- Shriner, S.A.; Root, J.J.; Lutman, M.W.; Kloft, J.M.; VanDalen, K.K.; Sullivan, H.J.; White, T.S.; Milleson, M.P.; Hairston, J.L.; Chandler, S.C.; et al. Surveillance for Highly Pathogenic H5 Avian Influenza Virus in Synanthropic Wildlife Associated with Poultry Farms during an Acute Outbreak. Sci. Rep. 2016, 6, 36237. [Google Scholar] [CrossRef]
- Alkie, T.N.; Cox, S.; Embury-Hyatt, C.; Stevens, B.; Pople, N.; Pybus, M.J.; Xu, W.; Hisanaga, T.; Suderman, M.; Koziuk, J.; et al. Characterization of Neurotropic HPAI H5N1 Viruses with Novel Genome Constellations and Mammalian Adaptive Mutations in Free-Living Mesocarnivores in Canada. Emerg. Microbes Infect. 2023, 12, 2186608. [Google Scholar] [CrossRef]
- Bordes, L.; Vreman, S.; Heutink, R.; Roose, M.; Venema, S.; Pritz-Verschuren, S.B.E.; Rijks, J.M.; Gonzales, J.L.; Germeraad, E.A.; Engelsma, M.; et al. Highly Pathogenic Avian Influenza H5N1 Virus Infections in Wild Red Foxes (Vulpes vulpes) Show Neurotropism and Adaptive Virus Mutations. Microbiol. Spectr. 2023, 11, e0286722. [Google Scholar] [CrossRef]
- Caliendo, V.; Leijten, L.; van de Bildt, M.W.G.; Fouchier, R.A.M.; Rijks, J.M.; Kuiken, T. Pathology and Virology of Natural Highly Pathogenic Avian Influenza H5N8 Infection in Wild Common Buzzards (Buteo buteo). Sci. Rep. 2022, 12, 920. [Google Scholar] [CrossRef]
- Hill, N.J.; Ma, E.J.; Meixell, B.W.; Lindberg, M.S.; Boyce, W.M.; Runstadler, J.A.; Bahl, J.; Krauss, S.; Kühnert, D.; Fourment, M.; et al. Influenza A Virus Migration and Persistence in North American Wild Birds. PLoS Pathog. 2013, 9, e1003570. [Google Scholar]
- Prosser, D.J.; Chen, J.; Ahlstrom, C.A.; Reeves, A.B.; Poulson, R.L.; Sullivan, J.D.; McAuley, D.; Callahan, C.R.; McGowan, P.C.; Bahl, J.; et al. Maintenance and Dissemination of Avian-Origin Influenza A Virus within the Northern Atlantic Flyway of North America. PLoS Pathog. 2022, 18, e1010605. [Google Scholar] [CrossRef]
- Huang, Z.Y.X.; Xu, C.; van Langevelde, F.; Ma, Y.; Langendoen, T.; Mundkur, T.; Si, Y.; Tian, H.; Kraus, R.H.S.; Gilbert, M.; et al. Contrasting Effects of Host Species and Phylogenetic Diversity on the Occurrence of HPAI H5N1 in European Wild Birds. J. Anim. Ecol. 2019, 88, 1044–1053. [Google Scholar] [CrossRef]
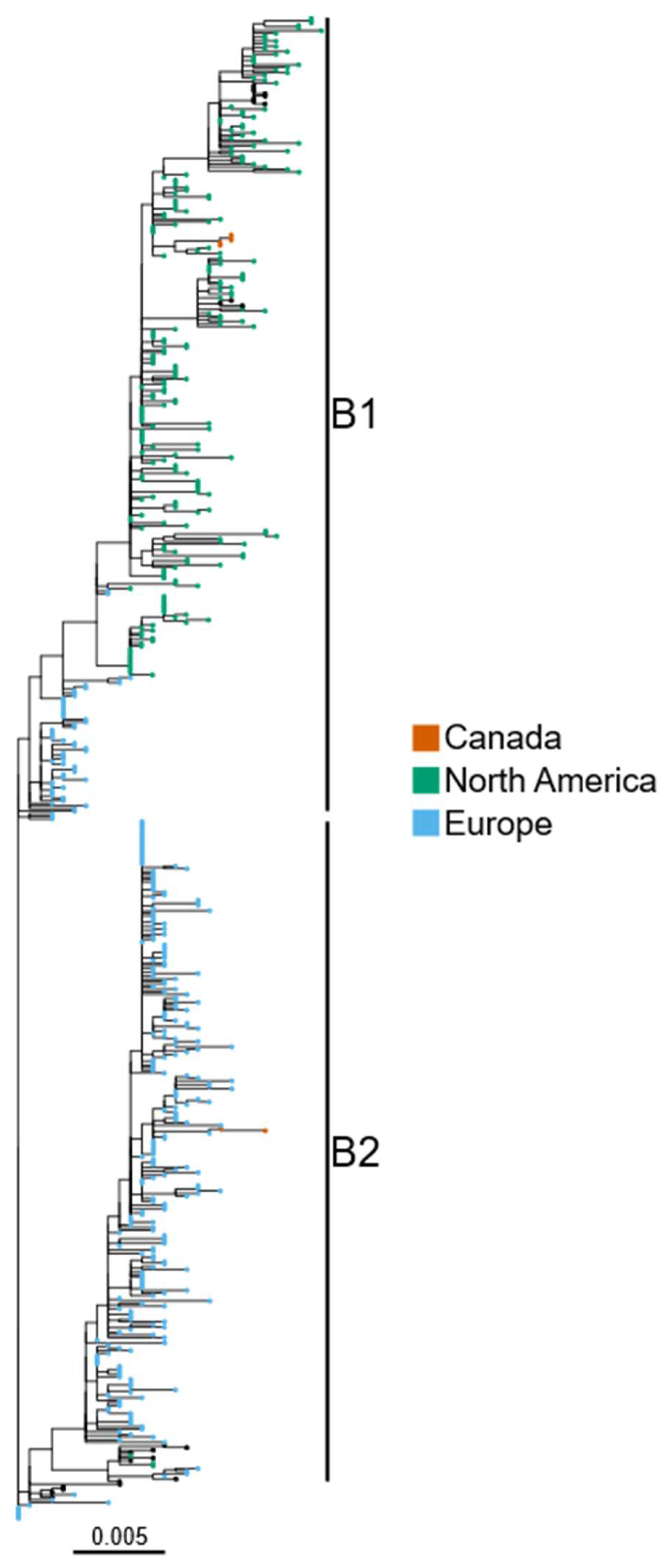

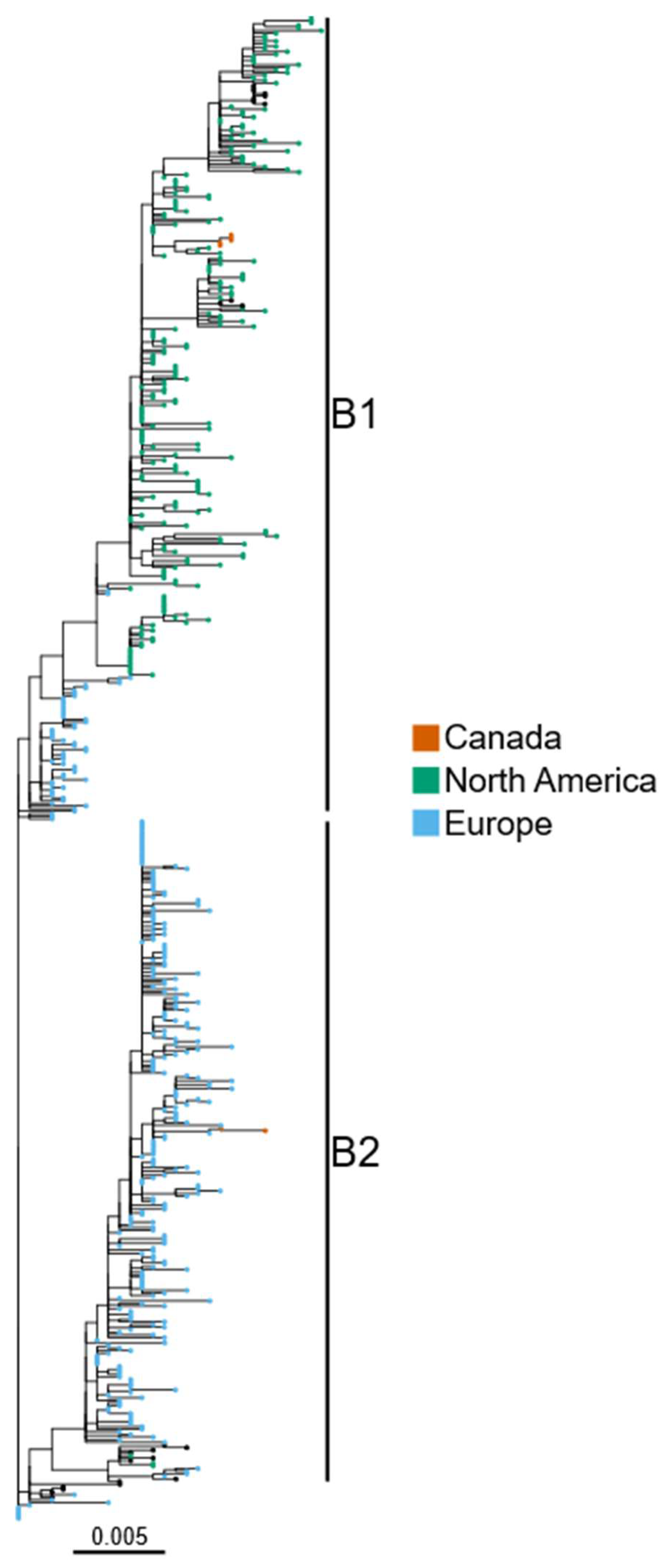{kind=link}
| Sequences | Collection Date | Species | Location | Phylogenetic Group | GISAID EpiFlu Accession Number |
|---|---|---|---|---|---|
| A/Red fox/NL/FAV-0075/2023 | 31 January 2023 | Red fox | Newfoundland | 1 | EPI_ISL_17494318 |
| A/American crow/PEI/FAV-0019-01/2022 | 26 December 2022 | American crow | Prince Edward Island | 1 | EPI_ISL_17494319 |
| A/American crow/PEI/FAV-0019-02/2022 | 28 December 2022 | American crow | Prince Edward Island | 2 | EPI_ISL_17478907 |
| A/American crow/PEI/FAV-0019-03/2022 | 28 December 2022 | American crow | Prince Edward Island | 2 | EPI_ISL_17479113 |
| A/American crow/PEI/FAV-0019-04/2022 | 28 December 2022 | American crow | Prince Edward Island | 2 | EPI_ISL_17479167 |
| A/American crow/PEI/FAV-0019-05/2022 | 28 December 2022 | American crow | Prince Edward Island | 2 | EPI_ISL_17479251 |
| A/American crow/PEI/FAV-0019-06/2022 | 28 December 2022 | American crow | Prince Edward Island | 2 | EPI_ISL_17479396 |
| Segment | Group 1 | Group 2 | ||||
|---|---|---|---|---|---|---|
| Node Date | Lower Bound | Higher Bound | Node Date | Lower Bound | Higher Bound | |
| PB2 | 22 December 2022 | 12 October 2022 | 26 December 2022 | 24 August 2022 | 2 June 2022 | 31 October 2022 |
| PB1 | 12 October 2022 | 6 August 2022 | 12 October 2022 | 28 December 2022 | 18 October 2022 | 28 December 2022 |
| PA | 9 November 2022 | 22 September 2022 | 20 December 2022 | 29 October 2022 | 10 September 2022 | 2 December 2022 |
| HA | 28 October 2022 | 28 August 2022 | 22 December 2022 | 28 December 2022 | 2 November 2022 | 28 December 2022 |
| NP | 12 October 2022 | 12 July 2022 | 12 October 2022 | 31 August 2022 | 11 June 2022 | 11 November 2022 |
| NA | 24 November 2022 | 27 August 2022 | 21 December 2022 | 22 June 2022 | 15 March 2022 | 6 October 2022 |
| MP | 26 December 2022 | 23 July 2022 | 26 December 2022 | 2 February 2022 | 11 September 2021 | 12 August 2022 |
| NS | 10 October 2022 | 4 May 2022 | 12 October 2022 | 27 July 2022 | 26 March 2022 | 27 November 2022 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alkie, T.N.; Byrne, A.M.P.; Jones, M.E.B.; Mollett, B.C.; Bourque, L.; Lung, O.; James, J.; Yason, C.; Banyard, A.C.; Sullivan, D.; et al. Recurring Trans-Atlantic Incursion of Clade 2.3.4.4b H5N1 Viruses by Long Distance Migratory Birds from Northern Europe to Canada in 2022/2023. Viruses 2023, 15, 1836. https://doi.org/10.3390/v15091836
Alkie TN, Byrne AMP, Jones MEB, Mollett BC, Bourque L, Lung O, James J, Yason C, Banyard AC, Sullivan D, et al. Recurring Trans-Atlantic Incursion of Clade 2.3.4.4b H5N1 Viruses by Long Distance Migratory Birds from Northern Europe to Canada in 2022/2023. Viruses. 2023; 15(9):1836. https://doi.org/10.3390/v15091836
Chicago/Turabian StyleAlkie, Tamiru N., Alexander M. P. Byrne, Megan E. B. Jones, Benjamin C. Mollett, Laura Bourque, Oliver Lung, Joe James, Carmencita Yason, Ashley C. Banyard, Daniel Sullivan, and et al. 2023. "Recurring Trans-Atlantic Incursion of Clade 2.3.4.4b H5N1 Viruses by Long Distance Migratory Birds from Northern Europe to Canada in 2022/2023" Viruses 15, no. 9: 1836. https://doi.org/10.3390/v15091836
APA StyleAlkie, T. N., Byrne, A. M. P., Jones, M. E. B., Mollett, B. C., Bourque, L., Lung, O., James, J., Yason, C., Banyard, A. C., Sullivan, D., Signore, A. V., Lang, A. S., Baker, M., Dawe, B., Brown, I. H., & Berhane, Y. (2023). Recurring Trans-Atlantic Incursion of Clade 2.3.4.4b H5N1 Viruses by Long Distance Migratory Birds from Northern Europe to Canada in 2022/2023. Viruses, 15(9), 1836. https://doi.org/10.3390/v15091836