
Genetic Variation between Asian and Mediterranean Populations of Cucurbit Aphid-Borne Yellows Virus




Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. RNA Extraction, RT-PCR
2.3. Recombination Analysis
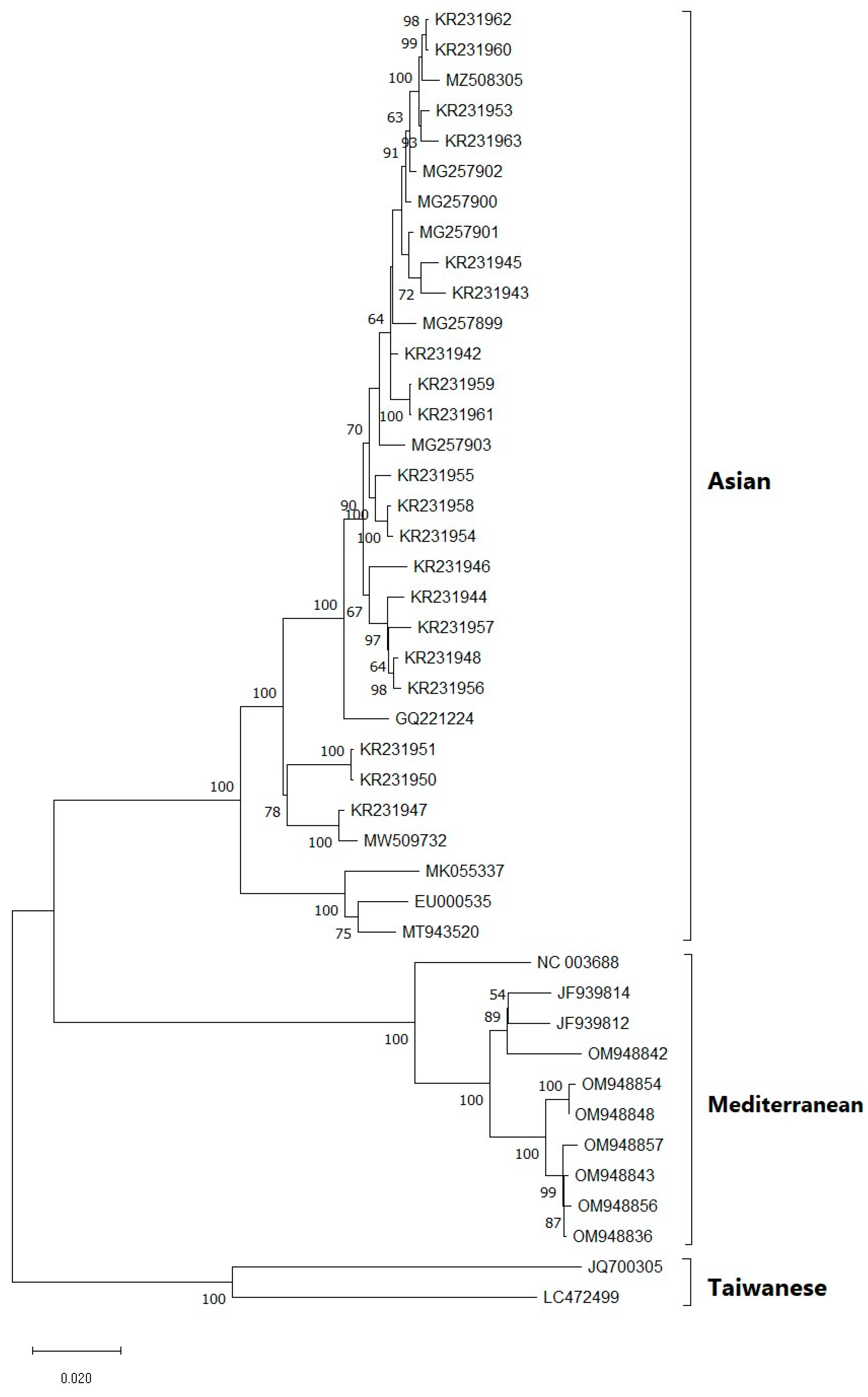
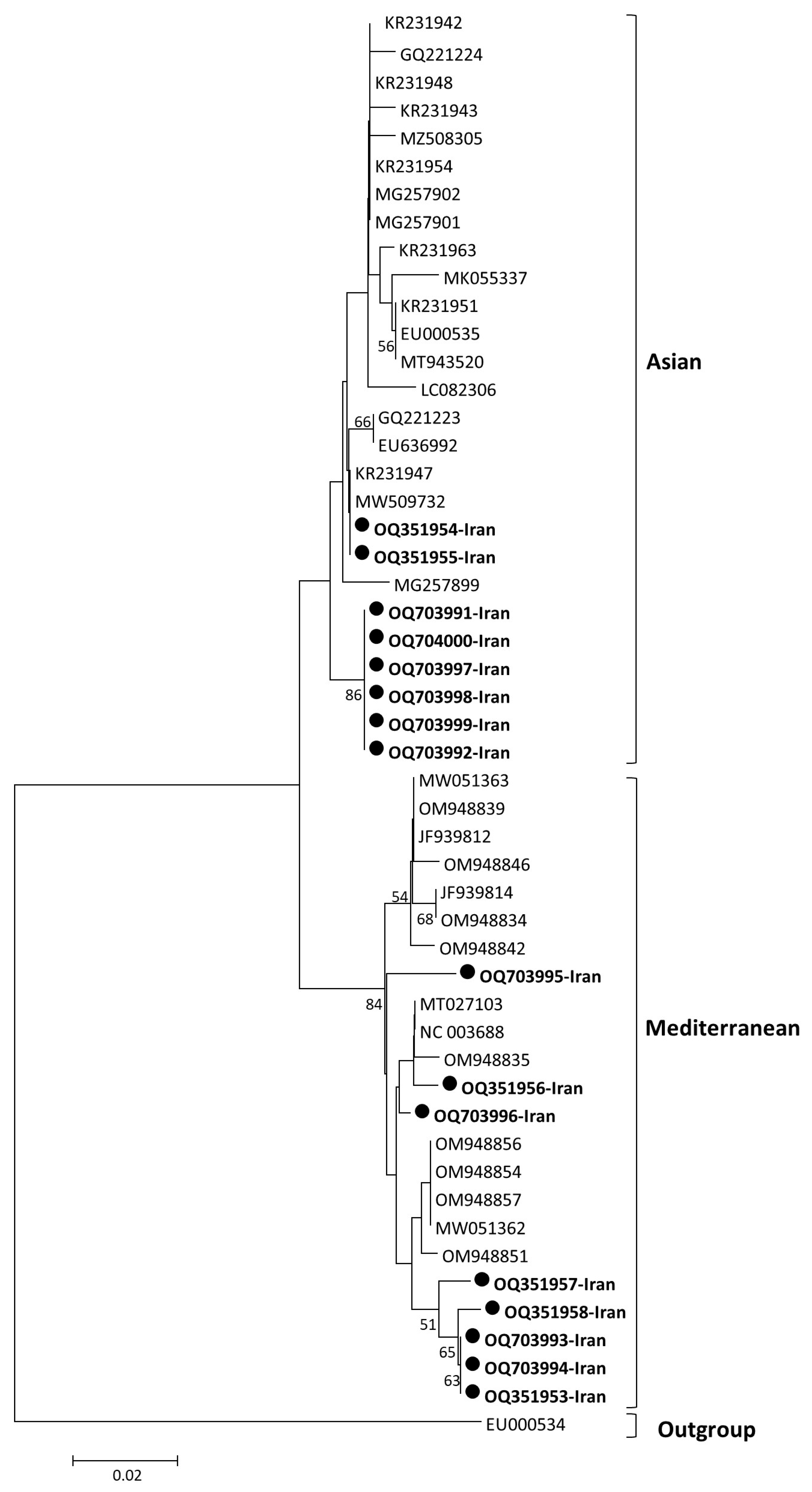
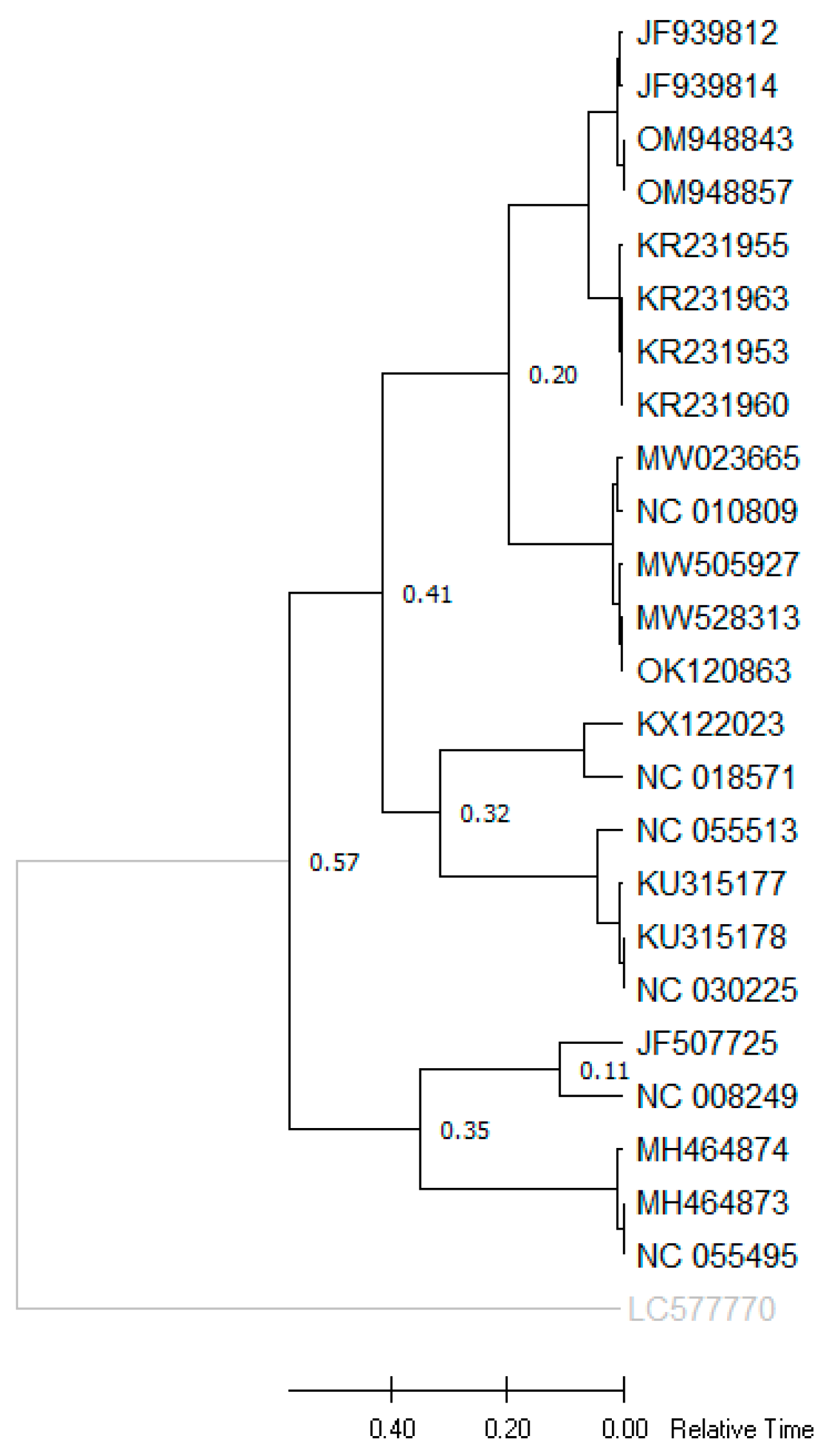
2.4. Phylogenetic Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Country | Isolates/strains | Host | Accession Number | Collection Date |
|---|---|---|---|---|
| South Korea (30) | SW1, SW2, CY3, C-HS1, WM-YS10, NW2, GS2, CY6, NW5, GM16, HS1,GS6, HD118, HD1, GS1, SW25, SW1(14), HS2, NW18, GM7, NW1, NW2(14), SW64, CY4,melon-GJ, Cumber-AS-kr1, M-CY31, M-BY1, C-AS1, K1 | melon, melon, melon, cucumber, watermelon, melon, melon, melon, melon, melon, melon, melon, melon, melon, melon, melon, melon, melon, melon, melon, melon, melon, melon, melon, melon, cucumber, melon, melon, cucumber, melon | KR231959, KR231961, KR231942, MG257900, MG257903, KR231955, KR231948, KR231944, KR231957, KR231946, KR231952, KR231949, KR231951, KR231950, KR231947, KR231962, KR231960, KR231953, KR231958, KR231945, KR231954, KR231956, KR231963, KR231943, MW509732, MZ508305, MG257902, MG257901, MG257899, LC082306 | 2014, 2014, 2013, 2016, 2016, 2013, 2014, 2014, 2013, 2013, 2014, 2014, 2014,2014, 2014, 2014, 2014, 2014, 2014, 2013, 2014, 2014, 2014, 2013, 2020, 2021, 2016, 2016, 2016, 2014 |
| Japan: Okayama (1) | CABYV-JAN | Cucumber | GQ221224 | - |
| China (5) | CABYV-FJ, -, -, CABYV-CZ, CABYV-QY | Squash, cantaloupe, cucurbit, zucchini, suakwa vegetable sponge | GQ221223, EU636992, EU000535, HQ439023, MT943520 | -, -, -, -, 2017 |
| USA (1) | BL-4 | Pumpkin | MK055337 | 2017 |
| Spain (29) | Sq/2004/1.9, Sq/2003/7.2, MEC-12, LP63, 64.2M/M, 9.2Z/A, 9.1M/M, 9.1M/M, 8.1M/M, 8.1Me/A, 7.1M/M, 67.1Z/M, 59.1M/A, 53.1M/M, 3.2M/M, 23.1M/M, 22.1Z/M, 2.1Z/M, 2.1M/M, 2.1M/M, 19.1Z/A, 19.1M/M, 19.1M/M, 18.1Z/A, 18.1M/A, 11.1Z/M, 10.1Z/M, 1.1M/M, Sq/2005/9.2 | -, -, melon, watermelon, melon, zucchini, melon, melon, melon, melon, melon, zucchini, melon, melon, melon, melon, zucchini, zucchini, melon, melon, zucchini, melon, melon, zucchini, melon, zucchini, zucchini, melon, - | JF939814, JF939812, MW051362, MW051363, OM948857, OM948856, OM948855, OM948854, OM948853, OM948852, OM948851, OM948850, OM948849, OM948848, OM948847, OM948846, OM948845, OM948844, OM948843, OM948842, OM948841, OM948840, OM948839, OM948838, OM948837, OM948836, OM948835, OM948834, JF939813 | 2017, -, -, 2014, 2019, 2018, 2013, 2019, 2014, 2016, 2014, 2013, 2018, 2020, 2020, 2017, 2015, 2019, 2015, 2013, 2011, 2019, 2019, 2015, 2015, 2016, 2012, 2020, 2014, - |
| Papua New Guinea (1) | 10PN | cucumber | MG780352 | 2016 |
| Taiwan (2) | CABYV-R-TW82, CABYV-C-TW20 | Luffa aegyptiaca, Momordica charantia | JQ700306, JQ700305 | 2009, 2008 |
| France (1) | EM160093 | melon | MT027103 | 2016 |
| Indonesia (1) | - | cucumber | LC472499 | 2017 |
| India (2) | POL-WM, POL-SQ | watermelon, squash | MN688220, MN688219 |
2.5. Population Genetic Parameters
2.6. Molecular Dating Analysis
3. Results
3.1. Field Observation
3.2. PCR Amplification
3.3. Recombination Isolates
3.4. Phylogenetic Analysis
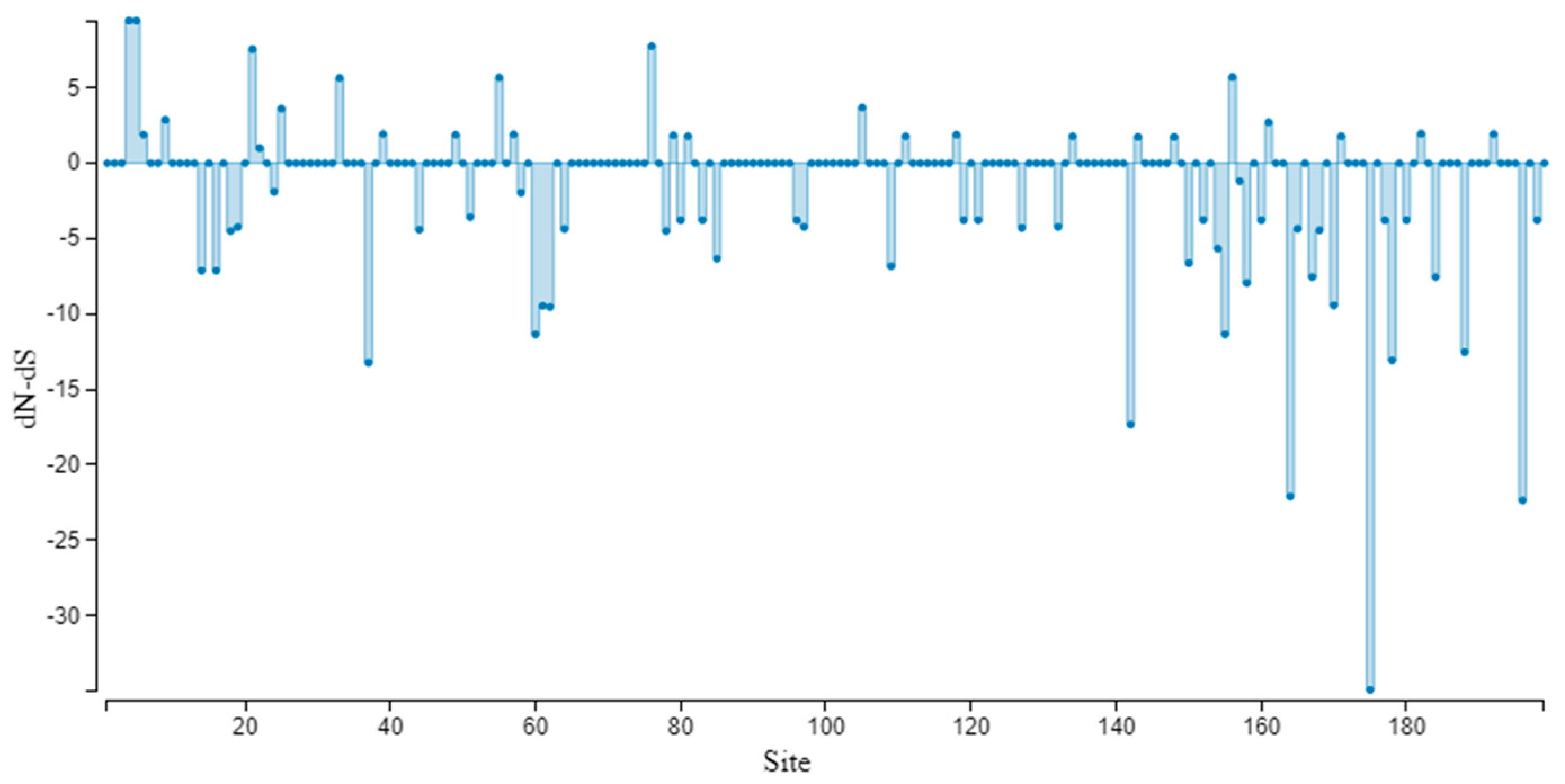
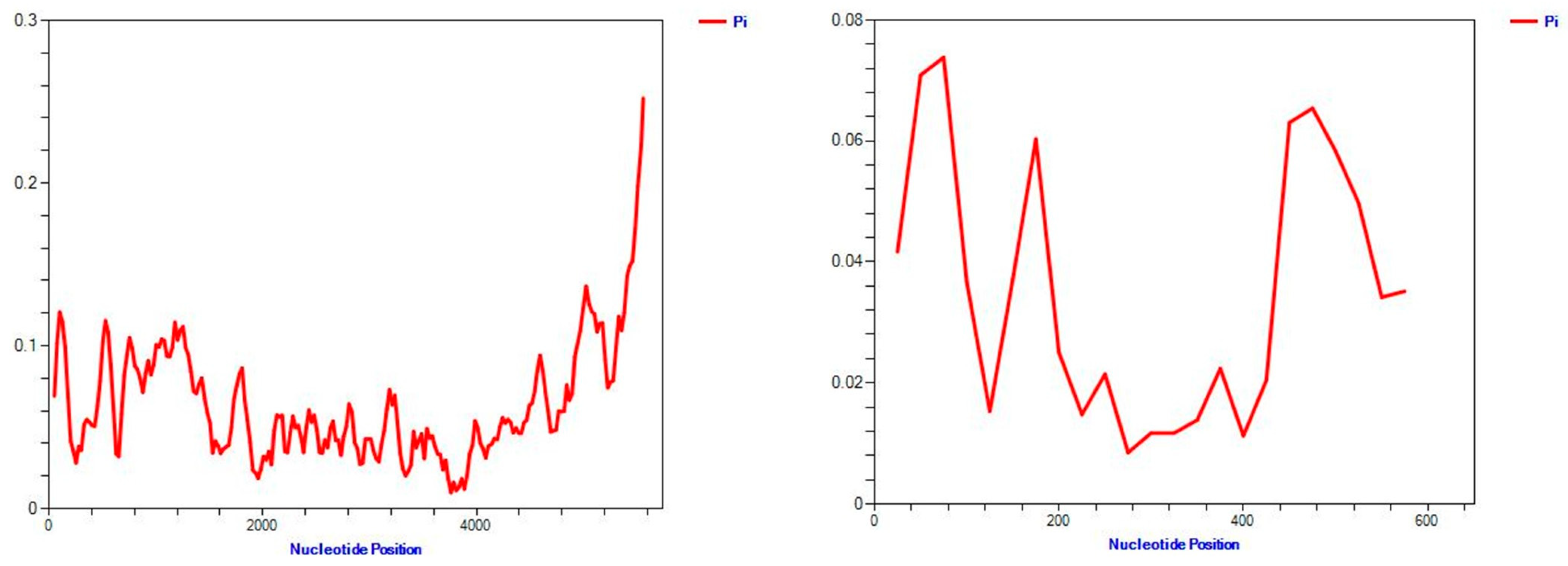
3.5. Genetic Variation and Selection Pressure Analyses
3.6. Neutrality Test
3.7. Genetic Differentiation
3.8. Divergence Time Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sastry, K.S.; Zitter, T.A. Management of Virus and Viroid Diseases of Crops in the Tropics. In Plant Virus and Viroid Diseases in the Tropics: Volume 2: Epidemiology and Management; Springer: Dordrecht, The Netherlands, 2014; pp. 149–480. [Google Scholar]
- Abkhoo, J. Serological and molecular detection and prevalence of Cucurbit aphid-borne yellows virus in the Sistan region, Iran. Afr. J. Biotechnol. 2012, 11, 13119–13122. [Google Scholar]
- Morteza, F.; Nematollahi, S.; Koolivand, D. Detection and molecular characterization of two Potyvirus species on cucurbits from Northwestern of Iran. J. Genet. Resour. 2021, 7, 106–115. [Google Scholar] [CrossRef]
- Nematollahi, S.; Panahborhani, N.; Koolivand, D. Molecular characterization and population evolution analysis of Watermelon mosaic virus isolates on cucurbits of Northwest Iran. 3 Biotech 2021, 11, 43. [Google Scholar] [CrossRef]
- Pilar Rabadán, M.; Juárez, M.; De Moya-Ruiz, C.; Gómez, P. Aphid-borne viruses infecting cultivated watermelon and squash in Spain: Characterization of a variant of cucurbit aphidborne yellows virus (CABYV). Plant Pathol. 2021, 70, 1476–1485. [Google Scholar] [CrossRef]
- Sõmera, M.; Fargette, D.; Hébrard, E.; Sarmiento, C. ICTV Virus Taxonomy Profile: Solemoviridae. J. Gen. Virol. 2021, 102, 001707. [Google Scholar] [CrossRef]
- Radeva-Ivanova, V.P.; Pasev, G.; Lyall, R.; Angelov, M.; Nankar, A.; Kostova, D. First report of Cucurbit aphid-borne yellows virus causing yellowing disease on pumpkin (Cucurbita pepo) and cucumber (Cucumis sativus) in Bulgaria. Plant Dis. 2022, 106, 2538. [Google Scholar] [CrossRef]
- Lecoq, H.; Bourdin, D.; Wipf-Scheibel, C.; Bon, M.; Lot, H.; Lemaire, O.; Herrbach, E. A new yellowing disease of cucurbits caused by a Luteovirus, cucurbit aphid-borne yellows virus. Plant Pathol. 1992, 41, 749–761. [Google Scholar] [CrossRef]
- Radouane, N.; Ezrari, S.; Belabess, Z.; Tahiri, A.; Tahzima, R.; Massart, S.; Jijakli, H.; Benjelloun, M.; Lahlali, R. Viruses of cucurbit crops: Current status in the Mediterranean Region. Phytopathol. Mediterr. 2021, 60, 493–519. [Google Scholar] [CrossRef]
- Maina, S.; Barbetti, M.J.; Edwards, O.R.; Minemba, D.; Areke, M.W.; Jones, R.A.C. First complete genome sequence of Cucurbit aphid-borne yellows virus from Papua New Guinea. Genome Announc. 2018, 6, e00162-18. [Google Scholar] [CrossRef]
- Krishnan, N.; Kumari, S.; Pandey, K.K.; Pandey, S.; Behera, T.K.; Gandhi, K.; Kenyon, L. Molecular characterization of novel Cucurbit aphid borne yellows virus strains infecting squash and watermelon in India. Physiol. Mol. Plant Pathol. 2022, 120, 101840. [Google Scholar] [CrossRef]
- Kassem, M.A.; Juarez, M.; Gómez, P.; Mengual, C.M.; Sempere, R.N.; Plaza, M.; Elena, S.F.; Moreno, A.; Fereres, A.; Aranda, M.A. Genetic diversity and potential vectors and reservoirs of Cucurbit aphid-borne yellows virus in southeastern Spain. Phytopathology 2013, 103, 1188–1197. [Google Scholar] [CrossRef]
- Kwak, H.-R.; Lee, H.J.; Kim, E.-A.; Seo, J.-K.; Kim, C.-S.; Lee, S.G.; Kim, J.-S.; Choi, H.-S.; Kim, M. Complete genome sequences and evolutionary analysis of Cucurbit aphid-borne yellows virus isolates from melon in Korea. Plant Pathol. 2018, 34, 532. [Google Scholar] [CrossRef]
- Vidal, A.H.; Lacorte, C.; Sanches, M.M.; Alves-Freitas, D.M.T.; Abreu, E.F.M.; Pinheiro-Lima, B.; Rosa, R.C.C.; Jesus, O.N.; Campos, M.A.; Felix, G.P.; et al. Characterization of Cucurbit Aphid-Borne Yellows Virus (CABYV) from Passion Fruit in Brazil: Evidence of a Complex of Species within CABYV Isolates. Viruses 2023, 15, 410. [Google Scholar] [CrossRef]
- Moya-Ruiz, C.D.; Gómez, P.; Juárez, M. Occurrence, Distribution, and Management of Aphid-Transmitted Viruses in Cucurbits in Spain. Pathogens 2023, 12, 422. [Google Scholar] [CrossRef]
- Costa, T.M.; Inoue-Nagata, A.K.; Vidal, A.H.; Ribeiro, S.d.G.; Nagata, T. The recombinant isolate of cucurbit aphid-borne yellows virus from Brazil is a polerovirus transmitted by whiteflies. Plant Pathol. 2020, 69, 1042–1050. [Google Scholar] [CrossRef]
- Vafaei, S.H.; Mahmoodi, M. Presence of recombinant strain of Cucurbit aphid borne yellows virus in Iran. Iran. J. Biotechnol. 2017, 15, 289. [Google Scholar] [CrossRef]
- Santosa, A.I.; Randa-Zelyüt, F.; Karanfil, A.; Korkmaz, S.; Hartono, S.; Ertunç, F. Phylogenetic and diversity analyses revealed that leek yellow stripevirus population consists of three types: S, L, and N. Virus Genes 2023, 59, 121–131. [Google Scholar] [CrossRef]
- Topkaya, Ş.; Çelik, A.; Santosa, A.I.; Jones, R.A.C. Molecular Analysis of the Global Population of Potato Virus S Redefines Its Phylogeny, and Has Crop Biosecurity Implications. Viruses 2023, 15, 1104. [Google Scholar] [CrossRef]
- Gambino, G.; Perrone, I.; Gribaudo, I. A rapid and effective method for RNA extraction from different tissues of grapevine and other woody plants. Phytochem Anal. 2008, 19, 520–525. [Google Scholar] [CrossRef]
- Khanal, V.; Ali, A. First Complete Genome Sequence of Cucurbit Aphid-Borne Yellows Virus from Pumpkin in the United States. Microbiol. Resour. Announc. 2019, 8, e01448-18. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Tamura, K. Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G + C-content biases. Mol. Biol. Evol. 1992, 9, 678–687. [Google Scholar] [CrossRef]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef]
- Fu, Y.X.; Li, W.H. Statistical tests of neutrality of mutations. Genetics 1993, 133, 693–709. [Google Scholar] [CrossRef]
- Hudson, R.R. A new statistic for detecting genetic differentiation. Genetics 2000, 155, 2011–2014. [Google Scholar] [CrossRef]
- Çelik, A.; Coşkan, S.; Morca, A.F.; Santosa, A.I.; Koolivand, D. Insight into Population Structure and Evolutionary Analysis of the Emerging Tomato Brown Rugose Fruit Virus. Plants 2022, 11, 3279. [Google Scholar] [CrossRef]
- Mello, B. Estimating TimeTrees with MEGA and the TimeTree resource. Mol. Biol. Evol. 2018, 35, 2334–2342. [Google Scholar] [CrossRef]
- Mnari-Hattab, M.; Gauthier, N.; Zouba, A. Biological and Molecular Characterization of the Cucurbit aphid-borne yellows virus Affecting Cucurbits in Tunisia. Plant Dis. 2009, 93, 1065–1072. [Google Scholar] [CrossRef] [PubMed]
- Maachi, A.; Donaire, L.; Hernando, Y.; Aranda, M.A. Genetic differentiation and migration fluxes of viruses from melon crops and crop edge weeds. J. Virol. 2022, 96, e00421-22. [Google Scholar] [CrossRef] [PubMed]






| Population | N | h | Hd | S | η | k | π | dS | dN | ω |
|---|---|---|---|---|---|---|---|---|---|---|
| Complete genome | 44 | 43 | 0.999 | 1575 | 1889 | 359.165 | 0.06459 | 0.14167 | 0.04046 | 0.2855 |
| Phylogroups (Complete Genome) | ||||||||||
| Asian | 31 | 31 | 1.000 | 802 | 843 | 142.168 | 0.02505 | 0.05880 | 0.01464 | 0.2489 |
| Mediterranean | 11 | 10 | 0.982 | 470 | 488 | 162.145 | 0.02860 | 0.07279 | 0.01495 | 0.2053 |
| ORF0 | 44 | 38 | 0.990 | 216 | 247 | 48.854 | 0.06814 | 0.10700 | 0.05564 | 0.5200 |
| ORF1 + 2 | 44 | 43 | 0.999 | 860 | 1000 | 187.080 | 0.05920 | 0.10641 | 0.04486 | 0.4215 |
| ORF4 | 50 | 26 | 0.960 | 24 | 25 | 6.167 | 0.02909 | 0.06467 | 0.01814 | 0.2805 |
| ORF3 + 5 | 44 | 42 | 0.998 | 573 | 710 | 132.185 | 0.06633 | 0.19262 | 0.02610 | 0.1354 |
| ORF3 (CP) | 50 | 26 | 0.960 | 24 | 25 | 6.167 | 0.02909 | 0.04990 | 0.02143 | 0.4294 |
| Phylogroups (new isolates) (Coat Protein) | ||||||||||
| Asian | 27 | 12 | 0.900 | 13 | 13 | 2.365 | 0.01115 | 0.03583 | 0.00621 | 0.1733 |
| Mediterranean | 23 | 14 | 0.945 | 12 | 12 | 3.036 | 0.01432 | 0.05017 | 0.02311 | 0.4606 |
| Population | Fu and Li’s D* | Fu and Li’s F* | Tajima’s D |
|---|---|---|---|
| Complete genome | −0.85487 ns | −0.92764 ns | −0.64287 ns |
| Phylogroup | |||
| Asian | −1.10647 ns | −1.37784 ns | −1.26911 ns |
| Mediterranean | 0.31512 ns | 0.22766 ns | −0.13000 ns |
| ORF3 (CP) | −1.02637 ns | −0.64835 ns | 0.34283 ns |
| Phylogroup | |||
| Asian | −1.49049 ns | −1.57070 ns | −1.00962 ns |
| Mediterranean | −0.25873 ns | −0.29129 ns | −0.22998 ns |
| ORF4 (MP) | −1.02637 ns | −0.64835 ns | 0.34283 ns |
| ORF0 | −0.84544 ns | −0.86372 ns | −0.51353 ns |
| ORF1 + 2 | −0.83674 ns | −0.93552 ns | −0.69127 ns |
| ORF3 + 5 | −0.95533 ns | −1.03074 ns | −0.70499 ns |
| Comparison | αKS* | αKST* | p Value | αZ* | p Value | Snn | p Value | βFST |
|---|---|---|---|---|---|---|---|---|
| Complete genome | ||||||||
| Phylogroup | ||||||||
| All (n = 44)/Asian (n = 31) | 5.1803 | 0.0002 | 0.422 ns | 6.9397 | 0.633 ns | 0.1733 | 1.000 ns | 0.104 |
| All (n = 44)/Mediterranean (n = 11) | 5.3621 | 0.0423 | 0.000 *** | 6.1228 | 0.000 *** | 0.6242 | 0.774 ns | 0.482 |
| Asian (n = 31)/Mediterranean (n = 11) | 4.7411 | 0.1243 | 0.000 *** | 5.2430 | 0.000 *** | 1.000 | 0.000 *** | 0.758 |
| ORF4 (CP) | ||||||||
| Phylogroup | ||||||||
| All (n = 50)/Asian (n = 27) | 1.5485 | 0.0566 | 0.000 *** | 6.9248 | 0.000 *** | 0.5202 | 0.656 ns | 0.243 |
| All (n = 50)/Mediterranean (n = 23) | 1.6342 | 0.0718 | 0.000 *** | 6.7496 | 0.000 *** | 0.5191 | 0.815 ns | 0.293 |
| Asian (n = 27)/Mediterranean (n = 23) | 1.1987 | 0.3256 | 0.000 *** | 5.4576 | 0.000 *** | 1.000 | 0.000 *** | 0.718 |
| Species | Mean Patristic Distance | Ratios of CABYV and Other Patristic Distances |
|---|---|---|
| CABYV | 0.20 | 1 |
| MABYV | 0.20 | 1 |
| SABYV | 0.32 | 1.6 |
| Pumpkin polerovirus | 0.32 | 1.6 |
| PABYV | 0.32 | 1.6 |
| CpCSV | 0.35 | 1.75 |
| FBPV-1 | 0.35 | 1.75 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pouraziz, P.; Yousefi, M.; Santosa, A.I.; Koolivand, D. Genetic Variation between Asian and Mediterranean Populations of Cucurbit Aphid-Borne Yellows Virus. Viruses 2023, 15, 1714. https://doi.org/10.3390/v15081714
Pouraziz P, Yousefi M, Santosa AI, Koolivand D. Genetic Variation between Asian and Mediterranean Populations of Cucurbit Aphid-Borne Yellows Virus. Viruses. 2023; 15(8):1714. https://doi.org/10.3390/v15081714
Chicago/Turabian StylePouraziz, Parastoo, Milad Yousefi, Adyatma Irawan Santosa, and Davoud Koolivand. 2023. "Genetic Variation between Asian and Mediterranean Populations of Cucurbit Aphid-Borne Yellows Virus" Viruses 15, no. 8: 1714. https://doi.org/10.3390/v15081714
APA StylePouraziz, P., Yousefi, M., Santosa, A. I., & Koolivand, D. (2023). Genetic Variation between Asian and Mediterranean Populations of Cucurbit Aphid-Borne Yellows Virus. Viruses, 15(8), 1714. https://doi.org/10.3390/v15081714