

SARS-CoV-2 Spike Protein Is Capable of Inducing Cell–Cell Fusions Independent from Its Receptor ACE2 and This Activity Can Be Impaired by Furin Inhibitors or a Subset of Monoclonal Antibodies

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Oligonucleotides and Expression Constructs
2.2. Cell Culture
2.3. Generation of 293T-DSP Cells through Lentivirus Transduction
2.4. Dual Split Protein Cell Membrane Fusion Assay (DSP Assay)
2.5. Pseudovirus Entry Assay
2.6. Indirect Immunofluorescence Analysis
2.7. Immunoblotting
2.8. Antibodies
3. Results
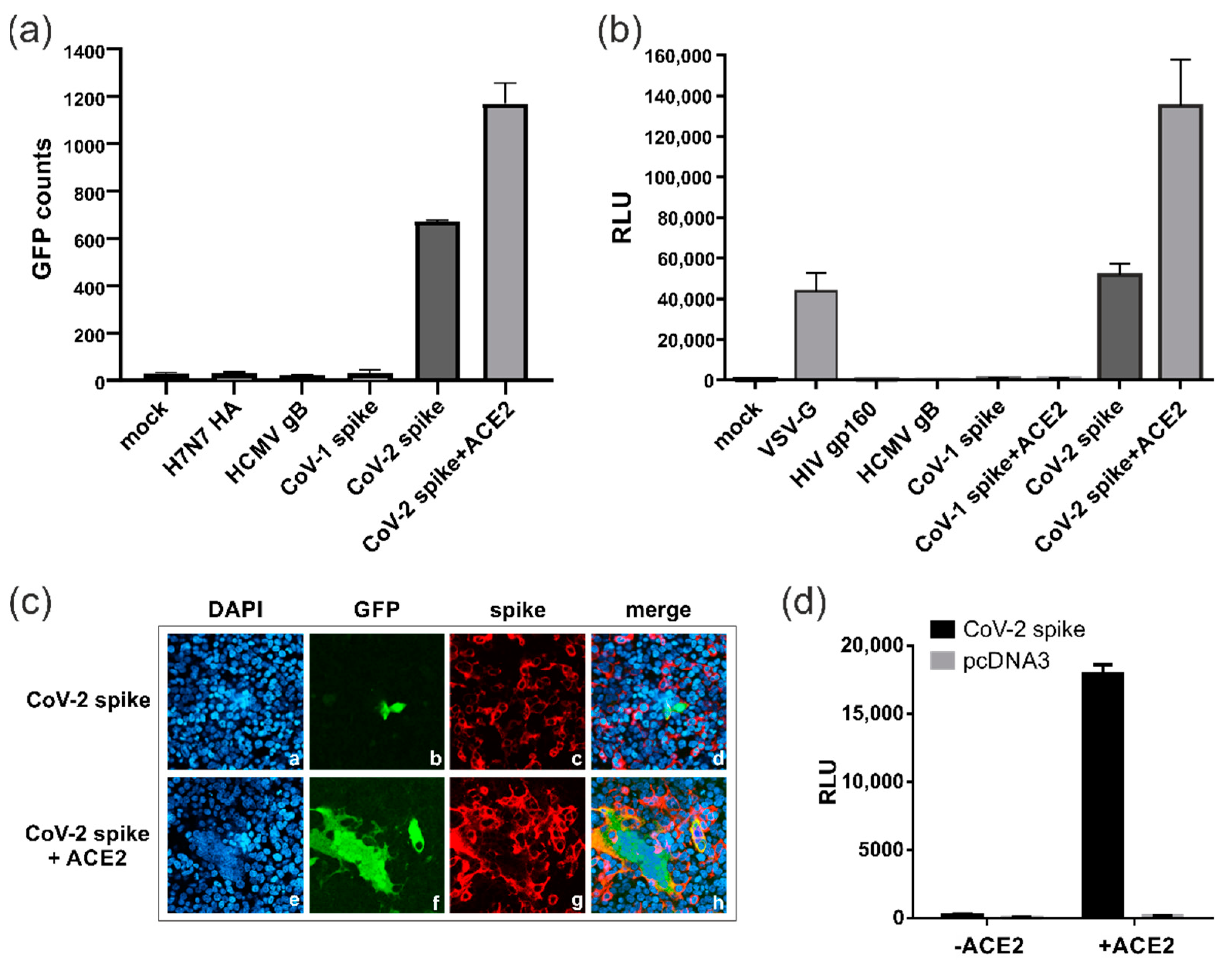
3.1. Establishment of a Dual Split Protein (DSP) Assay for Automated Quantification of Cell–Cell Fusions
3.2. SARS-CoV-2 Spike Protein Can Induce ACE2-Independent Cell–Cell Fusions
3.3. Proteolytic Cleavage of Spike by Furin but Not TMPRSS2 Is Essential for ACE2-Independent Cell–Cell Fusions
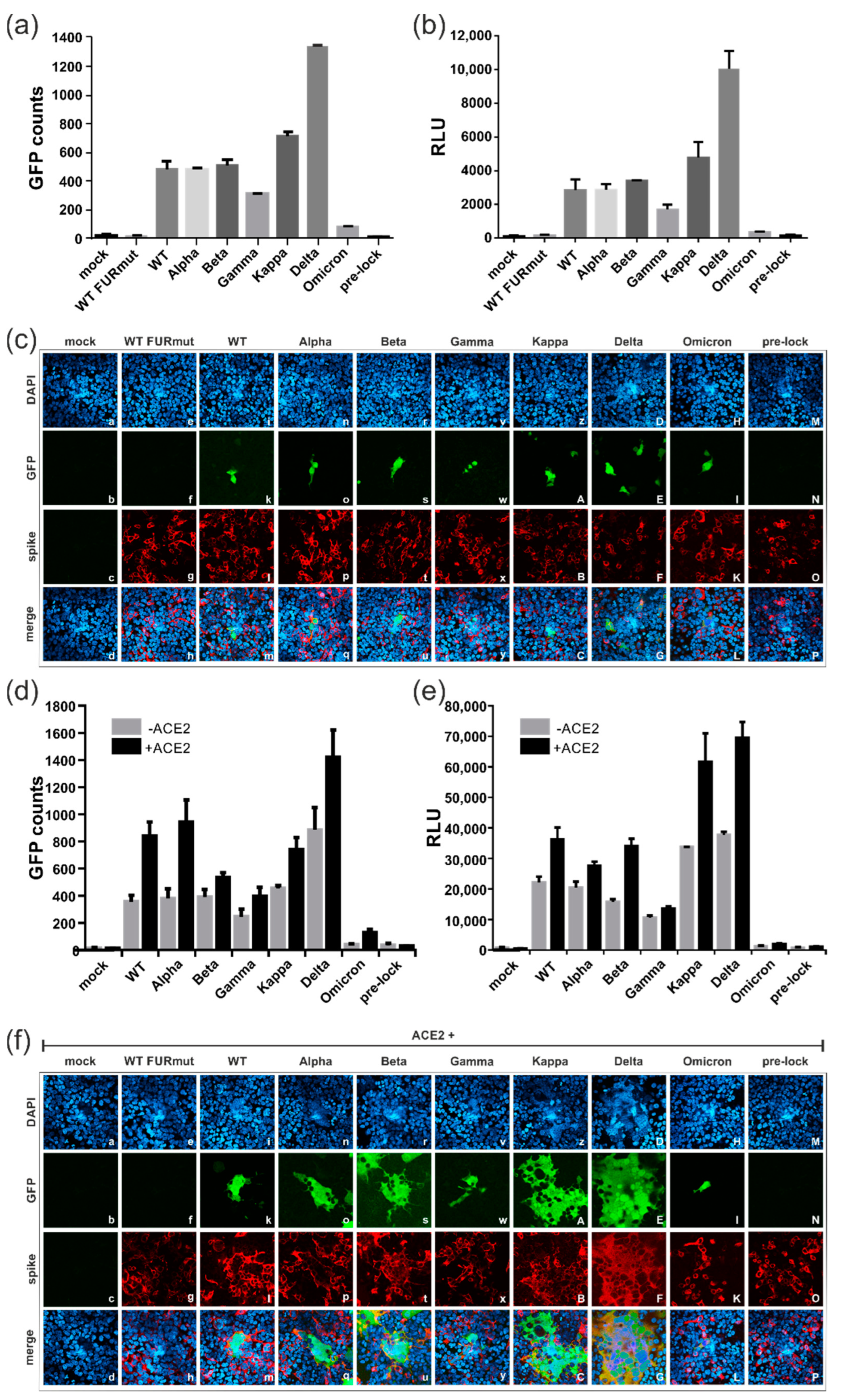
3.4. Spike Variants Greatly Differ in Their Capacity to Induce Cell Membrane Fusions
3.5. All Spike Variants Are Dependent on Furin Cleavage for Induction of ACE2-Independent Cell Fusions
3.6. Characterization of the ACE2-Independent Fusion Activity of Spike
3.7. Blocking of Spike-Induced Cell Fusions by Antibodies
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chan, J.F.; Kok, K.H.; Zhu, Z.; Chu, H.; To, K.K.; Yuan, S.; Yuen, K.Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef] [PubMed]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Pohlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784.e5. [Google Scholar] [CrossRef]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Coutard, B.; Valle, C.; de Lamballerie, X.; Canard, B.; Seidah, N.G.; Decroly, E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir. Res. 2020, 176, 104742. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904.e9. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Gomes, C.P.; Fernandes, D.E.; Casimiro, F.; da Mata, G.F.; Passos, M.T.; Varela, P.; Mastroianni-Kirsztajn, G.; Pesquero, J.B. Cathepsin L in COVID-19: From Pharmacological Evidences to Genetics. Front. Cell. Infect. Microbiol. 2020, 10, 589505. [Google Scholar] [CrossRef]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef]
- Takeda, M. Proteolytic activation of SARS-CoV-2 spike protein. Microbiol. Immunol. 2022, 66, 15–23. [Google Scholar] [CrossRef]
- Hempel, T.; Raich, L.; Olsson, S.; Azouz, N.P.; Klingler, A.M.; Hoffmann, M.; Pohlmann, S.; Rothenberg, M.E.; Noe, F. Molecular mechanism of inhibiting the SARS-CoV-2 cell entry facilitator TMPRSS2 with camostat and nafamostat. Chem. Sci. 2021, 12, 983–992. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.W.; Chao, T.L.; Li, C.L.; Chiu, M.F.; Kao, H.C.; Wang, S.H.; Pang, Y.H.; Lin, C.H.; Tsai, Y.M.; Lee, W.H.; et al. Furin Inhibitors Block SARS-CoV-2 Spike Protein Cleavage to Suppress Virus Production and Cytopathic Effects. Cell Rep. 2020, 33, 108254. [Google Scholar] [CrossRef]
- Zeng, C.; Evans, J.P.; King, T.; Zheng, Y.M.; Oltz, E.M.; Whelan, S.P.J.; Saif, L.J.; Peeples, M.E.; Liu, S.L. SARS-CoV-2 spreads through cell-to-cell transmission. Proc. Natl. Acad. Sci. USA 2022, 119, e2111400119. [Google Scholar] [CrossRef] [PubMed]
- Buchrieser, J.; Dufloo, J.; Hubert, M.; Monel, B.; Planas, D.; Rajah, M.M.; Planchais, C.; Porrot, F.; Guivel-Benhassine, F.; Van der Werf, S.; et al. Syncytia formation by SARS-CoV-2-infected cells. EMBO J. 2020, 39, e106267. [Google Scholar] [CrossRef] [PubMed]
- Rajah, M.M.; Bernier, A.; Buchrieser, J.; Schwartz, O. The Mechanism and Consequences of SARS-CoV-2 Spike-Mediated Fusion and Syncytia Formation. J. Mol. Biol. 2022, 434, 167280. [Google Scholar] [CrossRef] [PubMed]
- Bussani, R.; Schneider, E.; Zentilin, L.; Collesi, C.; Ali, H.; Braga, L.; Volpe, M.C.; Colliva, A.; Zanconati, F.; Berlot, G.; et al. Persistence of viral RNA, pneumocyte syncytia and thrombosis are hallmarks of advanced COVID-19 pathology. EBioMedicine 2020, 61, 103104. [Google Scholar] [CrossRef] [PubMed]
- Braga, L.; Ali, H.; Secco, I.; Chiavacci, E.; Neves, G.; Goldhill, D.; Penn, R.; Jimenez-Guardeno, J.M.; Ortega-Prieto, A.M.; Bussani, R.; et al. Drugs that inhibit TMEM16 proteins block SARS-CoV-2 spike-induced syncytia. Nature 2021, 594, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zheng, Y.; Niu, Z.; Zhang, B.; Wang, C.; Yao, X.; Peng, H.; Franca, D.N.; Wang, Y.; Zhu, Y.; et al. SARS-CoV-2 spike protein dictates syncytium-mediated lymphocyte elimination. Cell Death Differ. 2021, 28, 2765–2777. [Google Scholar] [CrossRef] [PubMed]
- Escalera, A.; Gonzalez-Reiche, A.S.; Aslam, S.; Mena, I.; Laporte, M.; Pearl, R.L.; Fossati, A.; Rathnasinghe, R.; Alshammary, H.; van de Guchte, A.; et al. Mutations in SARS-CoV-2 variants of concern link to increased spike cleavage and virus transmission. Cell Host Microbe 2022, 30, 373–387.e7. [Google Scholar] [CrossRef]
- Rajah, M.M.; Hubert, M.; Bishop, E.; Saunders, N.; Robinot, R.; Grzelak, L.; Planas, D.; Dufloo, J.; Gellenoncourt, S.; Bongers, A.; et al. SARS-CoV-2 Alpha, Beta, and Delta variants display enhanced Spike-mediated syncytia formation. EMBO J. 2021, 40, e108944. [Google Scholar] [CrossRef]
- Kondo, N.; Miyauchi, K.; Meng, F.; Iwamoto, A.; Matsuda, Z. Conformational changes of the HIV-1 envelope protein during membrane fusion are inhibited by the replacement of its membrane-spanning domain. J. Biol. Chem. 2010, 285, 14681–14688. [Google Scholar] [CrossRef]
- Reuter, N.; Kropff, B.; Schneiderbanger, J.K.; Alt, M.; Krawczyk, A.; Sinzger, C.; Winkler, T.H.; Britt, W.J.; Mach, M.; Thomas, M. Cell Fusion Induced by a Fusion-Active Form of Human Cytomegalovirus Glycoprotein B (gB) Is Inhibited by Antibodies Directed at Antigenic Domain 5 in the Ectodomain of gB. J. Virol. 2020, 94, e01276-20. [Google Scholar] [CrossRef]
- Peter, A.S.; Gruner, E.; Socher, E.; Fraedrich, K.; Richel, E.; Mueller-Schmucker, S.; Cordsmeier, A.; Ensser, A.; Sticht, H.; Uberla, K. Characterization of SARS-CoV-2 Escape Mutants to a Pair of Neutralizing Antibodies Targeting the RBD and the NTD. Int. J. Mol. Sci. 2022, 23, 8177. [Google Scholar] [CrossRef]
- Dispinseri, S.; Secchi, M.; Pirillo, M.F.; Tolazzi, M.; Borghi, M.; Brigatti, C.; De Angelis, M.L.; Baratella, M.; Bazzigaluppi, E.; Venturi, G.; et al. Neutralizing antibody responses to SARS-CoV-2 in symptomatic COVID-19 is persistent and critical for survival. Nat. Commun. 2021, 12, 2670. [Google Scholar] [CrossRef]
- Peter, A.S.; Roth, E.; Schulz, S.R.; Fraedrich, K.; Steinmetz, T.; Damm, D.; Hauke, M.; Richel, E.; Mueller-Schmucker, S.; Habenicht, K.; et al. A pair of noncompeting neutralizing human monoclonal antibodies protecting from disease in a SARS-CoV-2 infection model. Eur. J. Immunol. 2022, 52, 770–783. [Google Scholar] [CrossRef]
- Ohan, M.; David, L.; Peter, S.; Daniel, C. SARS-CoV-2 ANTIBODIES. Garvan Institute of Medical Research [AU], WO2022133545A1. 2022. Available online: https://patentimages.storage.googleapis.com/60/be/be/5a157de3d6c99e/WO2022133545A1.pdf (accessed on 14 May 2023).
- Hastie, E.; Cataldi, M.; Marriott, I.; Grdzelishvili, V.Z. Understanding and altering cell tropism of vesicular stomatitis virus. Virus Res. 2013, 176, 16–32. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, A.; Jungnickl, D.; Cordsmeier, A.; Peter, A.S.; Uberla, K.; Ensser, A. Cloning of a Passage-Free SARS-CoV-2 Genome and Mutagenesis Using Red Recombination. Int. J. Mol. Sci. 2021, 22, 10188. [Google Scholar] [CrossRef] [PubMed]
- Papa, G.; Mallery, D.L.; Albecka, A.; Welch, L.G.; Cattin-Ortola, J.; Luptak, J.; Paul, D.; McMahon, H.T.; Goodfellow, I.G.; Carter, A.; et al. Furin cleavage of SARS-CoV-2 Spike promotes but is not essential for infection and cell-cell fusion. PLoS Pathog. 2021, 17, e1009246. [Google Scholar] [CrossRef] [PubMed]
- Meng, B.; Abdullahi, A.; Ferreira, I.; Goonawardane, N.; Saito, A.; Kimura, I.; Yamasoba, D.; Gerber, P.P.; Fatihi, S.; Rathore, S.; et al. Altered TMPRSS2 usage by SARS-CoV-2 Omicron impacts infectivity and fusogenicity. Nature 2022, 603, 706–714. [Google Scholar] [CrossRef]
- Jiang, S.; Zhang, X.; Du, L. Therapeutic antibodies and fusion inhibitors targeting the spike protein of SARS-CoV-2. Expert Opin. Ther. Targets 2021, 25, 415–421. [Google Scholar] [CrossRef]
- Carsana, L.; Sonzogni, A.; Nasr, A.; Rossi, R.S.; Pellegrinelli, A.; Zerbi, P.; Rech, R.; Colombo, R.; Antinori, S.; Corbellino, M.; et al. Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: A two-centre descriptive study. Lancet Infect. Dis. 2020, 20, 1135–1140. [Google Scholar] [CrossRef] [PubMed]
- Rockx, B.; Kuiken, T.; Herfst, S.; Bestebroer, T.; Lamers, M.M.; Oude Munnink, B.B.; de Meulder, D.; van Amerongen, G.; van den Brand, J.; Okba, N.M.A.; et al. Comparative pathogenesis of COVID-19, MERS, and SARS in a nonhuman primate model. Science 2020, 368, 1012–1015. [Google Scholar] [CrossRef]
- Bryce, C.; Grimes, Z.; Pujadas, E.; Ahuja, S.; Beasley, M.B.; Albrecht, R.; Hernandez, T.; Stock, A.; Zhao, Z.; AlRasheed, M.R.; et al. Pathophysiology of SARS-CoV-2: The Mount Sinai COVID-19 autopsy experience. Mod. Pathol. 2021, 34, 1456–1467. [Google Scholar] [CrossRef]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef]
- Hofmann, H.; Pyrc, K.; van der Hoek, L.; Geier, M.; Berkhout, B.; Pohlmann, S. Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc. Natl. Acad. Sci. USA 2005, 102, 7988–7993. [Google Scholar] [CrossRef]
- Amraei, R.; Yin, W.; Napoleon, M.A.; Suder, E.L.; Berrigan, J.; Zhao, Q.; Olejnik, J.; Chandler, K.B.; Xia, C.; Feldman, J.; et al. CD209L/L-SIGN and CD209/DC-SIGN Act as Receptors for SARS-CoV-2. ACS Cent. Sci. 2021, 7, 1156–1165. [Google Scholar] [CrossRef] [PubMed]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef] [PubMed]
- Daly, J.L.; Simonetti, B.; Klein, K.; Chen, K.E.; Williamson, M.K.; Anton-Plagaro, C.; Shoemark, D.K.; Simon-Gracia, L.; Bauer, M.; Hollandi, R.; et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science 2020, 370, 861–865. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef]
- Gu, Y.; Cao, J.; Zhang, X.; Gao, H.; Wang, Y.; Wang, J.; He, J.; Jiang, X.; Zhang, J.; Shen, G.; et al. Receptome profiling identifies KREMEN1 and ASGR1 as alternative functional receptors of SARS-CoV-2. Cell Res. 2022, 32, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Nchioua, R.; Schundner, A.; Kmiec, D.; Prelli Bozzo, C.; Zech, F.; Koepke, L.; Graf, A.; Krebs, S.; Blum, H.; Frick, M.; et al. SARS-CoV-2 Variants of Concern Hijack IFITM2 for Efficient Replication in Human Lung Cells. J. Virol. 2022, 96, e0059422. [Google Scholar] [CrossRef]
- Shi, G.; Kenney, A.D.; Kudryashova, E.; Zani, A.; Zhang, L.; Lai, K.K.; Hall-Stoodley, L.; Robinson, R.T.; Kudryashov, D.S.; Compton, A.A.; et al. Opposing activities of IFITM proteins in SARS-CoV-2 infection. EMBO J. 2021, 40, e106501. [Google Scholar] [CrossRef]
- Zhao, X.; Zheng, S.; Chen, D.; Zheng, M.; Li, X.; Li, G.; Lin, H.; Chang, J.; Zeng, H.; Guo, J.T. LY6E Restricts Entry of Human Coronaviruses, Including Currently Pandemic SARS-CoV-2. J. Virol. 2020, 94, e00562-20. [Google Scholar] [CrossRef]
- Hallenberger, S.; Moulard, M.; Sordel, M.; Klenk, H.D.; Garten, W. The role of eukaryotic subtilisin-like endoproteases for the activation of human immunodeficiency virus glycoproteins in natural host cells. J. Virol. 1997, 71, 1036–1045. [Google Scholar] [CrossRef]
- Strive, T.; Borst, E.; Messerle, M.; Radsak, K. Proteolytic processing of human cytomegalovirus glycoprotein B is dispensable for viral growth in culture. J. Virol. 2002, 76, 1252–1264. [Google Scholar] [CrossRef]
- Stangherlin, L.M.; de Paula, F.N.; Icimoto, M.Y.; Ruiz, L.G.P.; Nogueira, M.L.; Braz, A.S.K.; Juliano, L.; da Silva, M.C.C. Positively Selected Sites at HCMV gB Furin Processing Region and Their Effects in Cleavage Efficiency. Front. Microbiol. 2017, 8, 934. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.; Leung, A.; Hisanaga, T.; Pickering, B.; Griffin, B.D.; Vendramelli, R.; Tailor, N.; Wong, G.; Bi, Y.; Babiuk, S.; et al. H7N9 Influenza Virus Containing a Polybasic HA Cleavage Site Requires Minimal Host Adaptation to Obtain a Highly Pathogenic Disease Phenotype in Mice. Viruses 2020, 12, 65. [Google Scholar] [CrossRef] [PubMed]
- Scheibner, D.; Ulrich, R.; Fatola, O.I.; Graaf, A.; Gischke, M.; Salaheldin, A.H.; Harder, T.C.; Veits, J.; Mettenleiter, T.C.; Abdelwhab, E.M. Variable impact of the hemagglutinin polybasic cleavage site on virulence and pathogenesis of avian influenza H7N7 virus in chickens, turkeys and ducks. Sci. Rep. 2019, 9, 11556. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, S.; Nagata, N.; Shirato, K.; Kawase, M.; Takeda, M.; Taguchi, F. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J. Virol. 2010, 84, 12658–12664. [Google Scholar] [CrossRef]
- Essalmani, R.; Jain, J.; Susan-Resiga, D.; Andreo, U.; Evagelidis, A.; Derbali, R.M.; Huynh, D.N.; Dallaire, F.; Laporte, M.; Delpal, A.; et al. Distinctive Roles of Furin and TMPRSS2 in SARS-CoV-2 Infectivity. J. Virol. 2022, 96, e0012822. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, L.; Wu, J.; Yu, Y.; Liu, S.; Li, T.; Li, Q.; Ding, R.; Wang, H.; Nie, J.; et al. A second functional furin site in the SARS-CoV-2 spike protein. Emerg. Microbes Infect. 2022, 11, 182–194. [Google Scholar] [CrossRef]
- Yu, S.; Zheng, X.; Zhou, B.; Li, J.; Chen, M.; Deng, R.; Wong, G.; Lavillette, D.; Meng, G. SARS-CoV-2 spike engagement of ACE2 primes S2′ site cleavage and fusion initiation. Proc. Natl. Acad. Sci. USA 2022, 119, e2111199119. [Google Scholar] [CrossRef]
- Molloy, S.S.; Thomas, L.; VanSlyke, J.K.; Stenberg, P.E.; Thomas, G. Intracellular trafficking and activation of the furin proprotein convertase: Localization to the TGN and recycling from the cell surface. EMBO J. 1994, 13, 18–33. [Google Scholar] [CrossRef]
- Vey, M.; Schafer, W.; Berghofer, S.; Klenk, H.D.; Garten, W. Maturation of the trans-Golgi network protease furin: Compartmentalization of propeptide removal, substrate cleavage, and COOH-terminal truncation. J. Cell Biol. 1994, 127, 1829–1842. [Google Scholar] [CrossRef]
- Zhang, J.; Xiao, T.; Cai, Y.; Lavine, C.L.; Peng, H.; Zhu, H.; Anand, K.; Tong, P.; Gautam, A.; Mayer, M.L.; et al. Membrane fusion and immune evasion by the spike protein of SARS-CoV-2 Delta variant. Science 2021, 374, 1353–1360. [Google Scholar] [CrossRef]
- Hu, B.; Chan, J.F.; Liu, H.; Liu, Y.; Chai, Y.; Shi, J.; Shuai, H.; Hou, Y.; Huang, X.; Yuen, T.T.; et al. Spike mutations contributing to the altered entry preference of SARS-CoV-2 omicron BA.1 and BA.2. Emerg. Microbes Infect. 2022, 11, 2275–2287. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Tomita, K.; Hirayama, Y.; Inoue, J.-I.; Kawaguchi, Y.; Gohda, J. SARS-CoV-2 Omicron spike H655Y mutation is responsible for enhancement of the endosomal entry pathway and reduction of cell surface entry pathways. BioRxiv 2022. [Google Scholar] [CrossRef]
- Chaguza, C.; Hahn, A.M.; Petrone, M.E.; Zhou, S.; Ferguson, D.; Breban, M.I.; Pham, K.; Pena-Hernandez, M.A.; Castaldi, C.; Hill, V.; et al. Accelerated SARS-CoV-2 intrahost evolution leading to distinct genotypes during chronic infection. Cell Rep. Med. 2023, 4, 100943. [Google Scholar] [CrossRef]
- Focosi, D.; Maggi, F. Recombination in Coronaviruses, with a Focus on SARS-CoV-2. Viruses 2022, 14, 1239. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lenoch, J.; Kohler, D.; DeLiberto, T.J.; Tang, C.Y.; Li, T.; Tao, Y.J.; Guan, M.; Compton, S.; Zeiss, C.; et al. SARS-CoV-2 Exposure in Norway Rats (Rattus norvegicus) from New York City. Mbio 2023, 14, e0362122. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Perez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef]
- Barrett, J.R.; Belij-Rammerstorfer, S.; Dold, C.; Ewer, K.J.; Folegatti, P.M.; Gilbride, C.; Halkerston, R.; Hill, J.; Jenkin, D.; Stockdale, L.; et al. Phase 1/2 trial of SARS-CoV-2 vaccine ChAdOx1 nCoV-19 with a booster dose induces multifunctional antibody responses. Nat. Med. 2021, 27, 279–288. [Google Scholar] [CrossRef]
- Mahallawi, W.H.; Mumena, W.A. Reactogenicity and Immunogenicity of the Pfizer and AstraZeneca COVID-19 Vaccines. Front. Immunol. 2021, 12, 794642. [Google Scholar] [CrossRef]
- Alhazmi, A.; Alamer, E.; Daws, D.; Hakami, M.; Darraj, M.; Abdelwahab, S.; Maghfuri, A.; Algaissi, A. Evaluation of Side Effects Associated with COVID-19 Vaccines in Saudi Arabia. Vaccines 2021, 9, 674. [Google Scholar] [CrossRef]
- Andrzejczak-Grzadko, S.; Czudy, Z.; Donderska, M. Side effects after COVID-19 vaccinations among residents of Poland. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 4418–4421. [Google Scholar]
- Omeish, H.; Najadat, A.; Al-Azzam, S.; Tarabin, N.; Abu Hameed, A.; Al-Gallab, N.; Abbas, H.; Rababah, L.; Rabadi, M.; Karasneh, R.; et al. Reported COVID-19 vaccines side effects among Jordanian population: A cross sectional study. Hum. Vaccin. Immunother. 2022, 18, 1981086. [Google Scholar] [CrossRef] [PubMed]
- Aldali, J.; Meo, S.A.; Al-Khlaiwi, T. Adverse Effects of Pfizer (BioNTech), Oxford-AstraZeneca (ChAdOx1 CoV-19), and Moderna COVID-19 Vaccines among the Adult Population in Saudi Arabia: A Cross-Sectional Study. Vaccines 2023, 11, 231. [Google Scholar] [CrossRef] [PubMed]
- Devi, K.P.; Pourkarim, M.R.; Thijssen, M.; Sureda, A.; Khayatkashani, M.; Cismaru, C.A.; Neagoe, I.B.; Habtemariam, S.; Razmjouei, S.; Khayat Kashani, H.R. A perspective on the applications of furin inhibitors for the treatment of SARS-CoV-2. Pharmacol. Rep. 2022, 74, 425–430. [Google Scholar] [CrossRef]
- Rahbar Saadat, Y.; Hosseiniyan Khatibi, S.M.; Zununi Vahed, S.; Ardalan, M. Host Serine Proteases: A Potential Targeted Therapy for COVID-19 and Influenza. Front. Mol. Biosci. 2021, 8, 725528. [Google Scholar] [CrossRef]
- Zhou, H.; Chen, Y.; Zhang, S.; Niu, P.; Qin, K.; Jia, W.; Huang, B.; Zhang, S.; Lan, J.; Zhang, L.; et al. Structural definition of a neutralization epitope on the N-terminal domain of MERS-CoV spike glycoprotein. Nat. Commun. 2019, 10, 3068. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lai, D.Y.; Zhang, H.N.; Jiang, H.W.; Tian, X.; Ma, M.L.; Qi, H.; Meng, Q.F.; Guo, S.J.; Wu, Y.; et al. Linear epitopes of SARS-CoV-2 spike protein elicit neutralizing antibodies in COVID-19 patients. Cell Mol. Immunol. 2020, 17, 1095–1097. [Google Scholar] [CrossRef]
- Poh, C.M.; Carissimo, G.; Wang, B.; Amrun, S.N.; Lee, C.Y.; Chee, R.S.; Fong, S.W.; Yeo, N.K.; Lee, W.H.; Torres-Ruesta, A.; et al. Two linear epitopes on the SARS-CoV-2 spike protein that elicit neutralising antibodies in COVID-19 patients. Nat. Commun. 2020, 11, 2806. [Google Scholar] [CrossRef]
- Ishikawa, H.; Meng, F.; Kondo, N.; Iwamoto, A.; Matsuda, Z. Generation of a dual-functional split-reporter protein for monitoring membrane fusion using self-associating split GFP. Protein Eng. Des. Sel. 2012, 25, 813–820. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer No. | Name | Sequence |
|---|---|---|
| 2-58 | 5AscI,H3,Kozakhu+muACE2 | GCATGGCGCGCCAAGCTTGCCACCATGTCMAGCTCYTCCTGGCTCCTTC |
| 2-59 | 3Pme1,NotSTOPhu+muACE2 | GCATGTTTAAACGCGGCCGCCTAAAAGGARGTCTGARCATCATC |
| 3-71 | cCoV-2 spike mutS1 | CCAGACACAGACAAACAGCCCCGCAGCGGCCGCATCTGTGGCCAGCCAGAGCATC |
| 3-72 | nCoV-2 spike mutS1 | GATGCTCTGGCTGGCCACAGATGCGGCCGCTGCGGGGCTGTTTGTCTGTGTCTGG |
| 3-73 | cCoV-2 spike mutS2′-AA | CGATCCTAGCAAGCCCAGCGCGGCGAGCTTCATCGAGGACCTGC |
| 3-74 | nCoV-2 spike mutS2′-AA | GCAGGTCCTCGATGAAGCTCGCCGCGCTGGGCTTGCTAGGATCG |
| 3-91 | 5mutSpike-K986P V987P | CGATATCCTGAGCAGACTGGACCCGCCGGAAGCCGAGGTGCAGATCGAC |
| 3-92 | 3mutSpike-K986P V987P | GTCGATCTGCACCTCGGCTTCCGGCGGGTCCAGTCTGCTCAGGATATCG |
| 4-11 | n dominant negative Furin | GCATTGTGGTCTCCATTCTGAACGATGGCATCGAG |
| 4-12 | c dominant negative Furin | CTCGATGCCATCGTTCAGAATGGAGACCACAATGC |
| 4-20 | cS2 Delta N679K+R681H | CCAGACACAGACAAAGAGCCACAGACGGGCC |
| 4-21 | nS2 Delta N679K+R681H | GGCCCGTCTGTGGCTCTTTGTCTGTGTCTGG |
| 4-28 | cS2 Omicron Y655H | GTCTGATCGGAGCCGAGCACGTGAACAATAGCTACG |
| 4-29 | nS2 Omicron Y655H | CGTAGCTATTGTTCACGTGCTCGGCTCCGATCAGAC |
| 4-30 | cS2 Delta H655Y | GTCTGATCGGAGCCGAGTACGTGAACAATAGCTACG |
| 4-31 | nS2 Delta H655Y | CGTAGCTATTGTTCACGTACTCGGCTCCGATCAGAC |
| 4-34 | cS2 Omicron K679N+H681R | CCAGACACAGACAAACAGCCGCAGACGGGCC |
| 4-35 | nS2 Omicron K679N+H681R | GGCCCGTCTGCGGCTGTTTGTCTGTGTCTGG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reuter, N.; Chen, X.; Kropff, B.; Peter, A.S.; Britt, W.J.; Mach, M.; Überla, K.; Thomas, M. SARS-CoV-2 Spike Protein Is Capable of Inducing Cell–Cell Fusions Independent from Its Receptor ACE2 and This Activity Can Be Impaired by Furin Inhibitors or a Subset of Monoclonal Antibodies. Viruses 2023, 15, 1500. https://doi.org/10.3390/v15071500
Reuter N, Chen X, Kropff B, Peter AS, Britt WJ, Mach M, Überla K, Thomas M. SARS-CoV-2 Spike Protein Is Capable of Inducing Cell–Cell Fusions Independent from Its Receptor ACE2 and This Activity Can Be Impaired by Furin Inhibitors or a Subset of Monoclonal Antibodies. Viruses. 2023; 15(7):1500. https://doi.org/10.3390/v15071500
Chicago/Turabian StyleReuter, Nina, Xiaohan Chen, Barbara Kropff, Antonia Sophia Peter, William J. Britt, Michael Mach, Klaus Überla, and Marco Thomas. 2023. "SARS-CoV-2 Spike Protein Is Capable of Inducing Cell–Cell Fusions Independent from Its Receptor ACE2 and This Activity Can Be Impaired by Furin Inhibitors or a Subset of Monoclonal Antibodies" Viruses 15, no. 7: 1500. https://doi.org/10.3390/v15071500
APA StyleReuter, N., Chen, X., Kropff, B., Peter, A. S., Britt, W. J., Mach, M., Überla, K., & Thomas, M. (2023). SARS-CoV-2 Spike Protein Is Capable of Inducing Cell–Cell Fusions Independent from Its Receptor ACE2 and This Activity Can Be Impaired by Furin Inhibitors or a Subset of Monoclonal Antibodies. Viruses, 15(7), 1500. https://doi.org/10.3390/v15071500