
Viral Infections and Schizophrenia: A Comprehensive Review
 ,
,  and
and 
Abstract
1. Introduction
1.1. The Disorder of Schizophrenia
1.2. Pathophysiology of Schizophrenia
Molecular Pathways in Schizophrenia
2. Risk Factors
2.1. Environmental Risk Factors
2.2. Pregnancy Complications
2.3. Viral Infections
2.3.1. Influenza Virus
2.3.2. Herpesviridae Family
Herpes Simplex Viruses (HSV)
Cytomegalovirus (CMV)
Epstein-Barr Virus (EBV)
2.3.3. Retroviruses
2.3.4. Borna Virus
2.3.5. Coronaviruses (CoVs)
2.3.6. Other Viral Infections
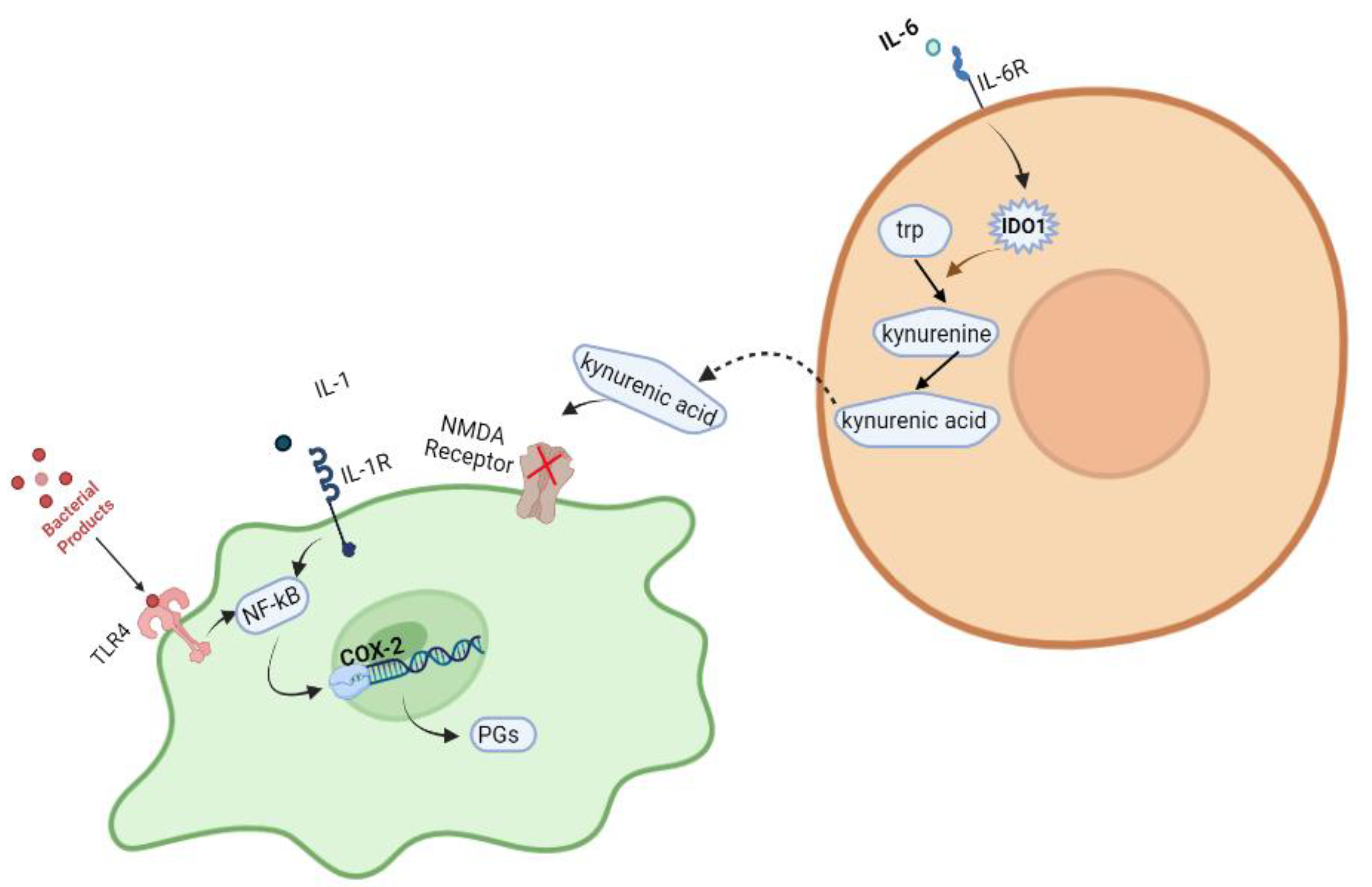
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lakhan, S.E.; Vieira, K.F. Schizophrenia pathophysiology: Are we any closer to a complete model? Ann. Gen. Psychiatry 2009, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Riedmuller, R.; Muller, S. Ethical Implications of the Mild Encephalitis Hypothesis of Schizophrenia. Front. Psychiatry 2017, 8, 38. [Google Scholar] [CrossRef]
- Vahia, V.N. Diagnostic and statistical manual of mental disorders 5: A quick glance. Indian J. Psychiatry 2013, 55, 220–223. [Google Scholar] [CrossRef]
- Wong, A.H.; Van Tol, H.H. Schizophrenia: From phenomenology to neurobiology. Neurosci. Biobehav. Rev. 2003, 27, 269–306. [Google Scholar] [CrossRef] [PubMed]
- Boog, G. Obstetrical complications and subsequent schizophrenia in adolescent and young adult offsprings: Is there a relationship? Eur. J. Obstet. Gynecol. Reprod. Biol. 2004, 114, 130–136. [Google Scholar] [CrossRef]
- Chung, Y.; Cannon, T.D. Brain imaging during the transition from psychosis prodrome to schizophrenia. J. Nerv. Ment. Dis. 2015, 203, 336–341. [Google Scholar] [CrossRef]
- Marcelis, M.; Navarro-Mateu, F.; Murray, R.; Selten, J.-P.; Van Os, J. Urbanization and psychosis: A study of 1942–1978 birth cohorts in The Netherlands. Psychol. Med. 1998, 28, 871–879. [Google Scholar] [CrossRef]
- Murray, R.M.; Lewis, S.W. Is schizophrenia a neurodevelopmental disorder? Br. Med. J. Clin. Res. Ed. 1987, 295, 681–682. [Google Scholar] [CrossRef]
- Bloom, F.E. Advancing a neurodevelopmental origin for schizophrenia. Arch. Gen. Psychiatry 1993, 50, 224–227. [Google Scholar] [CrossRef]
- Keshavan, M.S. Development, disease and degeneration in schizophrenia: A unitary pathophysiological model. J. Psychiatry Res. 1999, 33, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Pantelis, C.; Maruff, P. The cognitive neuropsychiatric approach to investigating the neurobiology of schizophrenia and other disorders. J. Psychosom. Res. 2002, 53, 655–664. [Google Scholar] [CrossRef]
- Tsuang, M. Schizophrenia: Genes and environment. Biol. Psychiatry 2000, 47, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.G. Schizophrenic birth seasonality in relation to the incidence of infectious diseases and temperature extremes. Arch. Gen. Psychiatry 1984, 41, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Zaidel, D.W.; Esiri, M.M.; Harrison, P.J. The hippocampus in schizophrenia: Lateralized increase in neuronal density and altered cytoarchitectural asymmetry. Psychol. Med. 1997, 27, 703–713. [Google Scholar] [CrossRef]
- Sanfilipo, M.; Lafargue, T.; Rusinek, H.; Arena, L.; Loneragan, C.; Lautin, A.; Wolkin, A. Volumetric measure of the frontal and temporal lobe regions in schizophrenia: Relationship to negative symptoms. Arch. Gen. Psychiatry 2000, 57, 471–480. [Google Scholar] [CrossRef]
- Thaker, G.K.; Carpenter, W.T., Jr. Advances in schizophrenia. Nat. Med. 2001, 7, 667–671. [Google Scholar] [CrossRef] [PubMed]
- Glavan, D.; Gheorman, V.; Gresita, A.; Hermann, D.M.; Udristoiu, I.; Popa-Wagner, A. Identification of transcriptome alterations in the prefrontal cortex, hippocampus, amygdala and hippocampus of suicide victims. Sci. Rep. 2021, 11, 18853. [Google Scholar] [CrossRef]
- Horvath, S.; Mirnics, K. Immune system disturbances in schizophrenia. Biol. Psychiatry 2014, 75, 316–323. [Google Scholar] [CrossRef]
- Khandaker, G.M.; Cousins, L.; Deakin, J.; Lennox, B.R.; Yolken, R.; Jones, P.B. Inflammation and immunity in schizophrenia: Implications for pathophysiology and treatment. Lancet Psychiatry 2014, 2, 258–270. [Google Scholar] [CrossRef]
- Brown, A.S.; Derkits, E.J. Prenatal Infection and Schizophrenia: A Review of Epidemiologic and Translational Studies. Am. J. Psychiatry 2010, 167, 261–280. [Google Scholar] [CrossRef]
- Miller, B.J.; Buckley, P.; Seabolt, W.; Mellor, A.; Kirkpatrick, B. Meta-analysis of cytokine alterations in schizophrenia: Clinical status and antipsychotic effects. Biol. Psychiatry 2011, 70, 663–671. [Google Scholar] [CrossRef]
- Mongan, D.; Ramesar, M.; Föcking, M.; Cannon, M.; Cotter, D. Role of inflammation in the pathogenesis of schizophrenia: A review of the evidence, proposed mechanisms and implications for treatment. Early Interv. Psychiatry 2020, 14, 385–397. [Google Scholar] [CrossRef]
- Massrali, A.; Adhya, D.; Srivastava, D.P.; Baron-Cohen, S.; Kotter, M.R. Virus-Induced Maternal Immune Activation as an Environmental Factor in the Etiology of Autism and Schizophrenia. Front Neurosci. 2022, 16, 834058. [Google Scholar] [CrossRef] [PubMed]
- Adell, A. Brain NMDA Receptors in Schizophrenia and Depression. Biomolecules 2020, 10, 947. [Google Scholar] [CrossRef]
- Giovanoli, S.; Engler, H.; Engler, A.; Richetto, J.; Voget, M.; Willi, R.; Winter, C.; Riva, M.A.; Mortensen, P.B.; Feldon, J.; et al. Stress in puberty unmasks latent neuropathological consequences of prenatal immune activation in mice. Science 2013, 339, 1095–1099. [Google Scholar] [CrossRef] [PubMed]
- Fields, J.K.; Gunther, S.; Sundberg, E.J. Structural Basis of IL-1 Family Cytokine Signaling. Front. Immunol. 2019, 10, 1412. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- MacDowell, K.S.; Pinacho, R.; Leza, J.C.; Costa, J.; Ramos, B.; García-Bueno, B. Differential regulation of the TLR4 signalling pathway in post-mortem prefrontal cortex and cerebellum in chronic schizophrenia: Relationship with SP transcription factors. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 79 Pt B, 481–492. [Google Scholar] [CrossRef]
- Beraki, S.; Aronsson, F.; Karlsson, H.; Ögren, S.O.; Kristensson, K. Influenza A virus infection causes alterations in expression of synaptic regulatory genes combined with changes in cognitive and emotional behaviors in mice. Mol. Psychiatry 2005, 10, 299–308. [Google Scholar] [CrossRef][Green Version]
- Eggers, A.E. A serotonin hypothesis of schizophrenia. Med. Hypotheses 2013, 80, 791–794. [Google Scholar] [CrossRef]
- Hensler, J.G.; Artigas, F.; Bortolozzi, A.; Daws, L.C.; De Deurwaerdère, P.; Milan, L.; Navailles, S.; Wouter, K. Catecholamine/Serotonin interactions: Systems thinking for brain function and disease. Adv. Pharmacol. 2013, 68, 167–197. [Google Scholar]
- Mueller, B.R.; Bale, T.L. Sex-specific programming of offspring emotionality after stress early in pregnancy. J. Neurosci. 2008, 28, 9055–9065. [Google Scholar] [CrossRef]
- Muller, N.; Schwarz, M.J. COX-2 inhibition in schizophrenia and major depression. Curr. Pharm. Des. 2008, 14, 1452–1465. [Google Scholar] [CrossRef]
- Delisi, L.; Angrist, A.O.; Bondy, B.; Davison, K.; Helmchen, H.; Tyrell, D.A.J. Nongenetic etiological factors group report. Biological perspectives of schizophrenia. Biol. Perspect. Schizophr. 2008, 6, 1054–1063. [Google Scholar]
- Benros, M.E.; Mortensen, P.B.; Eaton, W.W. Autoimmune diseases and infections as risk factors for schizophrenia. Ann. N. Y. Acad. Sci. 2012, 1262, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Hare, E.H.; Price, J.S.; Slater, E.; Guo, Z.-H.; Li, Z.-J.; Ma, Y.; Sun, J.; Guo, J.-H.; Li, W.-X.; Wang, Z.-Q.; et al. Schizophrenia and Season of Birth. Br. J. Psychiatry 1972, 120, 124–125. [Google Scholar] [CrossRef] [PubMed]
- Machon, R.A.; Mednick, S.A.; Schulsinger, F. The interaction of seasonality, place of birth, genetic risk and subsequent schizophrenia in a high risk sample. Br. J. Psychiatry 1983, 143, 383–388. [Google Scholar] [CrossRef]
- Pallast, E.G.; Jongbloet, P.H.; Straatman, H.M.; Zielhuis, G.A. Excess seasonality of births among patients with schizophrenia and seasonal ovopathy. Schizophr. Bull. 1994, 20, 269–276. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Torrey, E.F.; Miller, J.; Rawlings, R.; Yolken, R.H. Seasonality of births in schizophrenia and bipolar disorder: A review of the literature. Schizophr. Res. 1997, 28, 1–38. [Google Scholar] [CrossRef]
- Susser, E.S.; Brown, A.S.; Gorman, J.M. Prenatal Exposures in Schizophrenia; American Psychiatric Association: Washington, DC, USA, 1999. [Google Scholar]
- Pedersen, C.B.; Mortensen, P.B. Family history, place and season of birth as risk factors for schizophrenia in Denmark: A replication and reanalysis. Br. J. Psychiatry 2001, 179, 46–52. [Google Scholar] [CrossRef]
- Sham, P.C.; MacLean, C.J.; Kendler, K.S. Risk of schizophrenia and age difference with older siblings. Evidence for a maternal viral infection hypothesis? Br. J. Psychiatry 1993, 163, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Jaaskelainen, E.; Haapea, M.; Rautio, N.; Juola, P.; Penttilä, M.; Nordström, T.; Rissanen, I.; Husa, A.; Keskinen, E.; Marttila, R.; et al. Twenty Years of Schizophrenia Research in the Northern Finland Birth Cohort 1966: A Systematic Review. Schizophr. Res. Treat. 2015, 2015, 524875. [Google Scholar] [CrossRef] [PubMed]
- Delisi, L.E.; Smith, S.B.; Hamovit, J.R.; Maxwell, M.E.; Goldin, L.R.; Dingman, C.W.; Gershon, E.S. Herpes simplex virus, cytomegalovirus and Epstein-Barr virus antibody titres in sera from schizophrenic patients. Psychol. Med. 1986, 16, 757–763. [Google Scholar] [CrossRef]
- Brown, A.S.; Susser, E.S. Prenatal nutritional deficiency and risk of adult schizophrenia. Schizophr. Bull. 2008, 34, 1054–1063. [Google Scholar] [CrossRef] [PubMed]
- McNeil, T.F. Perinatal influences in the development of schizophrenia. Biol. Perspect. Schizophr. 1987, 125–138. [Google Scholar]
- Owen, M.J.; Lewis, S.W.; Murray, R.M. Obstetric complications and schizophrenia: A computed tomographic study. Psychol. Med. 1988, 18, 331–339. [Google Scholar] [CrossRef]
- Parnas, J.; Schulsinger, F.; Teasdale, T.W.; Feldman, P.M.; Mednick, S.A. Perinatal complications and clinical outcome within the schizophrenia spectrum. Br. J. Psychiatry 1982, 140, 416–420. [Google Scholar] [CrossRef]
- Jacobsen, B.; Kinney, D.K. Perinatal complications in adopted and non-adopted schizophrenics and their controls: Preliminary results. Acta Psychiatr. Scand. 1980, 62, 337–346. [Google Scholar] [CrossRef]
- Stilo, S.A.; Murray, R.M. The epidemiology of schizophrenia: Replacing dogma with knowledge. Dialogues Clin. Neurosci. 2022, 12, 305–315. [Google Scholar] [CrossRef]
- Woerner, M.G.; Pollack, M.; Klein, N.F. Birth weight and length in schizophrenics personality disorders and their siblings. Br. J. Psychiatry 1971, 118, 461–464. [Google Scholar] [CrossRef]
- Gilmore, J.H.; Murray, R.M. Prenatal and Perinatal Factors; American Psychiatric Publishing: Washington, DC, USA, 2006. [Google Scholar]
- Buka, S.L.; Tsuang, M.T.; Torrey, E.F.; Klebanoff, M.A.; Bernstein, D.; Yolken, R.H. Maternal Infections and Subsequent Psychosis Among Offspring. Arch. Gen. Psychiatry 2001, 58, 1032–1037. [Google Scholar] [CrossRef] [PubMed]
- Buka, S.L.; Tsuangabc, M.; Torreyd, E.F.; Klebanoff, M.A.; Wagner, R.L.; Yolken, R.H. Maternal Cytokine Levels during Pregnancy and Adult Psychosis. Brain Behav. Immun. 2001, 15, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Dammann, O.; Leviton, A. Infection remote from the brain, neonatal white matter damage, and cerebral palsy in the preterm infant. Semin. Pediatr. Neurol. 1998, 5, 190–201. [Google Scholar] [CrossRef]
- Mattei, D.; Djodari-Irani, A.; Hadar, R.; Pelz, A.; de Cossío, L.F.; Goetz, T.; Matyash, M.; Kettenmann, H.; Winter, C.; Wolf, A.S. Minocycline rescues decrease in neurogenesis, increase in microglia cytokines and deficits in sensorimotor gating in an animal model of schizophrenia. Brain Behav. Immun. 2014, 38, 175–184. [Google Scholar] [CrossRef]
- Rosenblat, J.D.; Cha, D.S.; Mansur, R.B.; McIntyre, R.S. Inflamed moods: A review of the interactions between inflammation and mood disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 53, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Estes, M.L.; McAllister, A.K. Maternal immune activation: Implications for neuropsychiatric disorders. Science 2016, 353, 772–777. [Google Scholar] [CrossRef]
- Fatemi, S.H.; Folsom, T.D. The neurodevelopmental hypothesis of schizophrenia, revisited. Schizophr. Bull. 2009, 35, 528–548. [Google Scholar] [CrossRef]
- Wright, P.; Takei, N.; Rifkin, L.; Murray, R.M. Maternal influenza, obstetric complications, and schizophrenia. Am. J. Psychiatry 1995, 152, 1714–1720. [Google Scholar]
- Akil, M.; Weinberger, D. Neuropathology and the neurodevelopmental model. Neuropathol. Schizophr. Prog. Interpret. 2000, 189–212. [Google Scholar] [CrossRef]
- Richart, S.M.; Simpson, S.A.; Krummenacher, C.; Whitbeck, J.C.; Pizer, L.I.; Cohen, G.H.; Eisenberg, R.J.; Wilcox, C.L. Entry of herpes simplex virus type 1 into primary sensory neurons in vitro is mediated by Nectin-1/HveC. J. Virol. 2003, 77, 3307–3311. [Google Scholar] [CrossRef]
- Ming, L. Influenza Virus Entry. In Advances in Experimental Medicine and Biology; Springer: Boston, MA, USA, 2012; Volume 726. [Google Scholar]
- Desforges, M.; Le Coupanec, A.; Dubeau, P.; Bourgouin, A.; Lajoie, L.; Dubé, M.; Talbot, P.J. Human Coronaviruses and Other Respiratory Viruses: Underestimated Opportunistic Pathogens of the Central Nervous System? Viruses 2019, 12, 14. [Google Scholar] [CrossRef]
- Mori, I.; Diehl, A.D.; Chauhan, A.; Ljunggren, H.G.; Kristensson, K. Selective targeting of habenular, thalamic midline and monoaminergic brainstem neurons by neurotropic influenza A virus in mice. J. Neurovirol. 1999, 5, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Li, X.-S.; Chen, L.; Chen, G.-Y.; Cheng, Y. A Network Analysis of Epigenetic and Transcriptional Regulation in a Neurodevelopmental Rat Model of Schizophrenia with Implicationwithr Translational Research. Schizophr. Bull. 2020, 46, 612–622. [Google Scholar] [CrossRef]
- Aguilar-Valles, A.; Rodrigue, B.; Matta-Camacho, E. Maternal Immune Activation and the Development of Dopaminergic Neurotransmission of the Offspring: Relevance for Schizophrenia and Other Psychoses. Front. Psychiatry 2020, 11, 852. [Google Scholar] [CrossRef]
- Mednick, S.A.; Machon, R.A.; Huttunen, M.O.; Bonett, D. Adult schizophrenia following prenatal exposure to an influenza epidemic. Arch. Gen. Psychiatry 1988, 45, 189–192. [Google Scholar] [CrossRef]
- Limosin, F.; Rouillon, F.; Payan, C.; Cohen, J.-M.; Strub, N. Prenatal exposure to influenza as a risk factor for adult schizophrenia. Acta Psychiatr. Scand. 2003, 107, 331–335. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan, E.; Gibson, T.; Colohan, H.A.; Walshe, D.; Buckley, P.; Larkin, C.; Waddington, J.L. Season of birth in schizophrenia. Evidence for confinement of an excess of winter births to patients without a family history of mental disorder. Br. J. Psychiatry 1991, 158, 764–769. [Google Scholar] [CrossRef]
- Brown, A.S.; Begg, M.D.; Gravenstein, S.; Schaefer, C.A.; Wyatt, R.J.; Bresnahan, M.; Babulas, V.P.; Susser, E.S. Serologic evidence of prenatal influenza in the etiology of schizophrenia. Arch. Gen. Psychiatry 2004, 61, 774–780. [Google Scholar] [CrossRef]
- Torrey, E.F.; Bowler, A.E.; Rawlings, R. An influenza epidemic and the seasonality of schizophrenic births. Psychiatry Biol. Factors 1991, 109–116. [Google Scholar]
- Burkhardt, E.; Berger, M.; Yolken, R.; Lin, A.; Yuen, H.; Wood, S.; Francey, S.; Thompson, A.; McGorry, P.; Nelson, B.; et al. Toxoplasma gondii, Herpesviridae and long-term risk of transition to first-episode psychosis in an ultra high-risk sample. Schizophr. Res. 2021, 233, 24–30. [Google Scholar] [CrossRef]
- Khandaker, G.M. Neuroinflammation and Schizophrenia; Jones, P.B., Ed.; Springer: Berlin/Heidelberg, Germany, 2020; Volume 44. [Google Scholar]
- Bray, P.F.; Bale, J.F.; Anderson, R.E.; Kern, E.R. Progressive neurological disease associated with chronic cytomegalovirus infection. Ann. Neurol. 1981, 9, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Steiner, I.; Kennedy, P.G.; Pachner, A.R. The neurotropic herpes viruses: Herpes simplex and varicella-zoster. Lancet Neurol. 2007, 6, 1015–1028. [Google Scholar] [CrossRef]
- Wang, Y.; Jia, J.; Wang, Y.; Li, F.; Song, X.; Qin, S.; Wang, Z.; Kitazato, K.; Wang, Y. Roles of HSV-1 infection-induced microglial immune responses in CNS diseases: Friends or foes? Crit. Rev. Microbiol. 2019, 45, 581–594. [Google Scholar] [CrossRef]
- Dickerson, F.B.; Boronow, J.J.; Stallings, C.; Origoni, A.E.; Ruslanova, I.; Yolken, R.H. Association of serum antibodies to herpes simplex virus 1 with cognitive deficits in individuals with schizophrenia. Arch. Gen. Psychiatry 2003, 60, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.; Bhatia, T.; Gauba, D.; Wood, J.; Long, C.; Prasad, K.; Dickerson, F.B.; Gur, R.E.; Gur, R.C.; Yolken, R.H.; et al. Exposure to herpes simplex virus, type 1 and reduced cognitive function. J. Psychiatr. Res. 2013, 47, 1680–1685. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yolken, R.H.; Torrey, E.F.; Lieberman, J.A.; Yang, S.; Dickerson, F.B. Serological evidence of exposure to Herpes Simplex Virus type 1 is associated with cognitive deficits in the CATIE schizophrenia sample. Schizophr. Res. 2011, 128, 61–65. [Google Scholar] [CrossRef]
- Mohagheghi, M.; Eftekharian, M.M.; Taheri, M.; Alikhani, M.Y. Determining the IgM and IgG antibodies titer against HSV1, HSV2 and CMV in the serum of schizophrenia patients. Hum. Antibodies 2018, 26, 87–93. [Google Scholar] [CrossRef]
- Mortensen, P.B.; Pedersen, C.B.; Hougaard, D.M.; Nørgaard-Petersen, B.; Mors, O.; Børglum, A.D.; Yolken, R.H. A Danish National Birth Cohort study of maternal HSV-2 antibodies as a risk factor for schizophrenia in their offspring. Schizophr. Res. 2010, 122, 257–263. [Google Scholar] [CrossRef]
- Severance, E.G.; Leister, F.; Lea, A.; Yang, S.; Dickerson, F.; Yolken, R.H. Complement C4 associations with altered microbial biomarkers exemplify gene-by-environment interactions in schizophrenia. Schizophr. Res. 2021, 234, 87–93. [Google Scholar] [CrossRef]
- Hanshaw, J.B. Congenital cytomegalovirus infection: A fifteen year perspective. J. Infect. Dis. 1971, 123, 555–561. [Google Scholar] [CrossRef]
- Bhumbra, N.A.; Nankervis, G.A. Cytomegalovirus infection. Overview and new developments. Postgrad Med. 1983, 73, 62–69. [Google Scholar] [CrossRef]
- Boppana, S.B.; Rivera, L.B.; Fowler, K.B.; Mach, M.; Britt, W.J. Intrauterine transmission of cytomegalovirus to infants of women with preconceptional immunity. N. Engl. J. Med. 2001, 344, 1366–1371. [Google Scholar] [CrossRef]
- Torrey, E.F.; Yolken, R.; Albrecht, P. Cytomegalovirus as a possible etiological agent in schizophrenia. Adv. Biol. Psychiatry 1983, 12, 150–160. [Google Scholar]
- Lucchese, G.; Flöel, A.; Stahl, B. A Peptide Link Between Human Cytomegalovirus Infection, Neuronal Migration, and Psychosis. Front. Psychiatry 2020, 11, 349. [Google Scholar] [CrossRef]
- Hoffmann, C.; Grossman, R.; Bokov, I.; Lipitz, S.; Biegon, A. Effect of cytomegalovirus infection on temporal lobe development in utero: Quantitative MRI studies. Eur. Neuropsychopharmacol. 2010, 20, 848–854. [Google Scholar] [CrossRef] [PubMed]
- Yolken, R.H.; Torrey, E.F. Are some cases of psychosis caused by microbial agents? A review of the evidence. Mol. Psychiatry 2008, 13, 470–479. [Google Scholar] [CrossRef]
- Kaufmann, C.A.; Weinberger, D.; Yolken, R.; Torrey, E.; Potkin, S. Viruses and schizophrenia. Lancet 1983, 2, 1136–1137. [Google Scholar] [CrossRef]
- Dickerson, F.; Kirkpatrick, B.; Boronow, J.; Stallings, C.; Origoni, A.; Yolken, R. Deficit schizophrenia: Association with serum antibodies to cytomegalovirus. Schizophr. Bull. 2006, 32, 396–400. [Google Scholar] [CrossRef]
- Albrecht, P.; Boone, E.; Torrey, E.F.; Hicks, J.; Daniel, N. Raised cytomegalovirus-antibody level in cerebrospinal fluid of schizophrenic patients. Lancet 1980, 2, 769–772. [Google Scholar] [CrossRef] [PubMed]
- Krech, U. Complement-fixing antibodies against cytomegalovirus in different parts of the world. Bull. World Health Organ. 1973, 49, 103–106. [Google Scholar]
- Lycke, E.; Norrby, R.; Roos, B.E. A serological study on mentally ill patients with particular reference to the prevalence of herpes virus infections. Br. J. Psychiatry 1974, 124, 273–279. [Google Scholar] [CrossRef]
- Jones-Brando, L.; Dickerson, F.; Ford, G.; Stallings, C.; Origoni, A.; Katsafanas, E.; Sweeney, K.; Squire, A.; Khushalani, S.; Yolken, R. Atypical immune response to Epstein-Barr virus in major depressive disorder. J. Affect. Disord. 2020, 264, 221–226. [Google Scholar] [CrossRef]
- Khandaker, G.M.; Stochl, J.; Zammit, S.; Lewis, G.; Jones, P.B. Childhood Epstein-Barr Virus infection and subsequent risk of psychotic experiences in adolescence: A population-based prospective serological study. Schizophr. Res. 2014, 158, 19–24. [Google Scholar] [CrossRef]
- Lemprière, S. Epstein–Barr virus and MS—A causal link. Nat. Rev. Neurol. 2022, 18, 128. [Google Scholar] [CrossRef]
- Dickerson, F.; Jones-Brando, L.; Ford, G.; Genovese, G.; Stallings, C.; Origoni, A.; O’dushlaine, C.; Katsafanas, E.; Sweeney, K.; Khushalani, S.; et al. Schizophrenia is Associated with an Aberrant Immune Response to Epstein-Barr Virus. Schizophr. Bull. 2019, 45, 1112–1119. [Google Scholar] [CrossRef] [PubMed]
- Gurling, H.M. Testing the retrovirus hypothesis of manic depression and schizophrenia with molecular genetic techniques. J. R. Soc. Med. 1988, 81, 332–334. [Google Scholar]
- Christensen, T. HERVs in neuropathogenesis. J. Neuroimmune. Pharmacol. 2010, 5, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Aftab, A.; Shah, A.A.; Hashmi, A.M. Pathophysiological Role of HERV-W in Schizophrenia. J. Neuropsychiatry Clin. Neurosci. 2016, 28, 17–25. [Google Scholar] [CrossRef]
- Huang, W.J.; Liu, Z.-C.; Wei, W.; Wang, G.-H.; Wu, J.-G.; Zhu, F. Human endogenous retroviral pol RNA and protein detected and identified in the blood of individuals with schizophrenia. Schizophr. Res. 2006, 83, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Li, S.; Hu, Y.; Yu, H.; Luo, F.; Zhang, Q.; Zhu, F. Implication of the env gene of the human endogenous retrovirus W family in the expression of BDNF and DRD3 and development of recent-onset schizophrenia. Schizophr. Bull. 2011, 37, 988–1000. [Google Scholar] [CrossRef]
- Yolken, R.H. Endogenous retroviruses and schizophrenia. Brain Res. Brain Res. Rev. 2000, 31, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Perron, H.; Mekaoui, L.; Bernard, C.; Veas, F.; Stefas, I.; Leboyer, M. Endogenous retrovirus type W GAG and envelope protein antigenemia in serum of schizophrenic patients. Biol. Psychiatry 2008, 64, 1019–1023. [Google Scholar] [CrossRef]
- Karlsson, H.; Bachmann, S.; Schröder, J.; McArthur, J.; Torrey, E.F.; Yolken, R.H. Retroviral RNA identified in the cerebrospinal fluids and brains of individuals with schizophrenia. Proc. Natl. Acad. Sci. USA 2001, 98, 4634–4639. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Schröder, J.; Nellåker, C.; Bottmer, C.; Bachmann, S.; Yolken, R.H.; Karlsson, H. Elevated levels of human endogenous retrovirus-W transcripts in blood cells from patients with first episode schizophrenia. Genes Brain Behav. 2008, 7, 103–112. [Google Scholar] [CrossRef]
- Gosztonyi, G.; Ludwig, H. Borna disease--neuropathology and pathogenesis. Curr. Top Microbiol. Immunol. 1995, 190, 39–73. [Google Scholar]
- Bilzer, T.; Planz, O.; Lipkin, W.I.; Stitz, L. Presence of CD4+ and CD8+ T cells and expression of MHC class I and MHC class II antigen in horses with Borna disease virus-induced encephalitis. Brain Pathol. 1995, 5, 223–230. [Google Scholar] [CrossRef]
- Ovanesov, M.V.; Ayhan, Y.; Wolbert, C.; Moldovan, K.; Sauder, C.; Pletnikov, M.V. Astrocytes play a key role in activation of microglia by persistent Borna disease virus infection. J. Neuroinflammation 2008, 5, 50. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.Y.; Zhang, F.-M.; Li, J.-H.; Li, G.-M.; Ma, P.-L.; Gu, H.-X.; Ikuta, K. Detection of Borna disease virus-p24 specific antibody in the sera of schizophrenic patients of China by means of Western-blot. Zhonghua Shi Yan He Lin Chuang Bing Du Xue Za Zhi. Chin. J. Exp. Clin. Virol. 2003, 17, 85–87. [Google Scholar]
- Iwata, Y.; Takahashi, K.; Peng, X.; Fukuda, K.; Ohno, K.; Ogawa, T.; Gonda, K.; Mori, N.; Niwa, S.; Shigeta, S. Detection and sequence analysis of borna disease virus p24 RNA from peripheral blood mononuclear cells of patients with mood disorders or schizophrenia and of blood donors. J. Virol. 1998, 72, 10044–10049. [Google Scholar] [CrossRef] [PubMed]
- Selten, J.P.; Slaets, J.; Kahn, R. Prenatal exposure to influenza and schizophrenia in Surinamese and Dutch Antillean immigrants to The Netherlands. Schizophr. Res. 1998, 30, 101–103. [Google Scholar] [CrossRef]
- Selten, J.; van Loon, A.M.; van Vliet, K.; Pleyte, W.; Hoek, H.; Kahn, R. Borna Disease Virus in Caribbean immigrants to the Netherlands, diagnosed with schizophrenia. Schizophr. Res. 1998, 1, 19. [Google Scholar] [CrossRef]
- Soltani, H.; Mohammadzadeh, S.; Makvandi, M.; Pakseresht, S.; Samarbaf-Zadeh, A. Detection of Borna Disease Virus (BDV) in Patients with First Episode of Schizophrenia. Iran J. Psychiatry 2016, 11, 257–261. [Google Scholar] [PubMed]
- Gioti, K.; Kottaridi, C.; Voyiatzaki, C.; Chaniotis, D.; Rampias, T.; Beloukas, A. Animal Coronaviruses Induced Apoptosis. Life 2021, 11, 185. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, A.; Youngblood, A.; Adnane, A.; Miller, B.J.; Goldsmith, D.R. Prenatal exposure to viral infection and neuropsychiatric disorders in offspring: A review of the literature and recommendations for the COVID-19 pandemic. Brain Behav. Immun. 2021, 91, 756–770. [Google Scholar] [CrossRef]
- Liu, J.; Li, Y.; Liu, Q.; Yao, Q.; Wang, X.; Zhang, H.; Wang, M.; Zhou, Y. SARS-CoV-2 cell tropism and multiorgan infection. Cell Discov. 2021, 7, 17. [Google Scholar] [CrossRef]
- Quincozes-Santos, A.; Rosa, R.L.; Tureta, E.F.; Bobermin, L.D.; Berger, M.; Guimarães, J.A.; Santi, L.; Beys-da-Silva, W.O. COVID-19 impacts the expression of molecular markers associated with neuropsychiatric disorders. Brain Behav. Immun. Health 2021, 11, 100196. [Google Scholar] [CrossRef]
- Lee, Y.H.; Kim, J.H.; Song, G.G. Pathway analysis of a genome-wide association study in schizophrenia. Gene 2013, 525, 107–115. [Google Scholar] [CrossRef]
- Yang, M.S.; Morris, D.W.; Donohoe, G.; Kenny, E.; O’Dushalaine, C.T.; Schwaiger, S.; Nangle, J.M.; Clarke, S.; Scully, P.; Quinn, J.; et al. Chitinase-3-like 1 (CHI3L1) gene and schizophrenia: Genetic association and a potential functional mechanism. Biol. Psychiatry 2008, 64, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Favre, G.; Lavenex, P.B.; Lavenex, P. Developmental regulation of expression of schizophrenia susceptibility genes in the primate hippocampal formation. Transl. Psychiatry 2012, 2, e173. [Google Scholar] [CrossRef] [PubMed]
- Kezurer, N.; Galron, D.; Golan, H.M. Increased susceptibility to mild neonatal stress in MTHFR deficient mice. Behav. Brain Res. 2013, 253, 240–252. [Google Scholar] [CrossRef]
- Takata, A.; Xu, B.; Ionita-Laza, I.; Roos, J.L.; Gogos, J.A.; Karayiorgou, M. Loss-of-function variants in schizophrenia risk and SETD1A as a candidate susceptibility gene. Neuron 2014, 82, 773–780. [Google Scholar] [CrossRef]
- Egbujo, C.N.; Sinclair, D.; Borgmann-Winter, K.E.; Arnold, S.E.; Turetsky, B.I.; Hahn, C.-G. Molecular evidence for decreased synaptic efficacy in the postmortem olfactory bulb of individuals with schizophrenia. Schizophr. Res. 2015, 168, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Severance, E.G.; Dickerson, F.B.; Viscidi, R.P.; Bossis, I.; Stallings, C.R.; Origoni, A.E.; Sullens, A.; Yolken, R.H. Coronavirus immunoreactivity in individuals with a recent onset of psychotic symptoms. Schizophr. Bull. 2011, 37, 101–107. [Google Scholar] [CrossRef]
- Zambrano, L.D.; Ellington, S.; Strid, P.; Galang, R.R.; Oduyebo, T.; Tong, V.T.; Woodworth, K.R.; Nahabedian, J.F.; Azziz-Baumgartner, E.; Gilboa, S.M.; et al. Update: Characteristics of Symptomatic Women of Reproductive Age with Laboratory-Confirmed SARS-CoV-2 Infection by Pregnancy Status—United States, 22 January–3 October 2020. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 1641–1647. [Google Scholar] [CrossRef]
- Lokken, E.M.; Taylor, G.G.; Huebner, E.M.; Vanderhoeven, J.; Hendrickson, S.; Coler, B.; Sheng, J.S.; Walker, C.L.; McCartney, S.A.; Kretzer, N.M.; et al. Higher severe acute respiratory syndrome coronavirus 2 infection rate in pregnant patients. Am. J. Obstet. Gynecol. 2021, 225, 75.e1–75.e16. [Google Scholar] [CrossRef] [PubMed]
- Sukhikh, G.; Petrova, U.; Prikhodko, A.; Starodubtseva, N.; Chingin, K.; Chen, H.; Bugrova, A.; Kononikhin, A.; Bourmenskaya, O.; Brzhozovskiy, A.; et al. Vertical Transmission of SARS-CoV-2 in Second Trimester Associated with Severe Neonatal Pathology. Viruses 2021, 13, 447. [Google Scholar] [CrossRef] [PubMed]
- Patane, L.; Morotti, D.; Giunta, M.R.; Sigismondi, C.; Piccoli, M.G.; Frigerio, L.; Mangili, G.; Arosio, M.; Cornolti, G. Vertical transmission of coronavirus disease 2019: Severe acute respiratory syndrome coronavirus 2 RNA on the fetal side of the placenta in pregnancies with coronavirus disease 2019-positive mothers and neonates at birth. Am. J. Obstet. Gynecol. MFM. 2020, 2, 100145. [Google Scholar] [CrossRef]
- Baud, D.; Greub, G.; Favre, G.; Gengler, C.; Jaton, K.; Dubruc, E.; Pomar, L. Second-Trimester Miscarriage in a Pregnant Woman With SARS-CoV-2 Infection. JAMA 2020, 323, 2198–2200. [Google Scholar] [CrossRef]
- Hoffman, M.C.; Freedman, R.; Law, A.J.; Clark, A.M.; Hunter, S.K. Maternal nutrients and effects of gestational COVID-19 infection on fetal brain development. Clin. Nutr. ESPEN 2021, 43, 1–8. [Google Scholar] [CrossRef]
- Hammond, C.J.; Hobbs, J.A. Parvovirus B19 infection of brain: Possible role of gender in determining mental illness and autoimmune thyroid disorders. Med. Hypotheses 2007, 69, 113–116. [Google Scholar] [CrossRef]
- Hobbs, J.A. Detection of adeno-associated virus 2 and parvovirus B19 in the human dorsolateral prefrontal cortex. J. Neurovirol. 2006, 12, 190–199. [Google Scholar] [CrossRef]
- Eagles, J.M. Are polioviruses a cause of schizophrenia? Br. J. Psychiatry 1992, 160, 598–600. [Google Scholar] [CrossRef]
- Squires, R.F. How a poliovirus might cause schizophrenia: A commentary on Eagles’ hypothesis. Neurochem. Res. 1997, 22, 647–656. [Google Scholar] [CrossRef]
- Joob, B.; Wiwanitkit, V. Zika Virus Outbreak, assisted reproduction patients and pregnancy. JBRA Assist. Reprod. 2018, 22, 75. [Google Scholar] [CrossRef]
- Pierson, T.C.; Diamond, M.S. The emergence of Zika virus and its new clinical syndromes. Nature 2018, 560, 573–581. [Google Scholar] [CrossRef]
- Elgueta, D.; Murgas, P.; Riquelme, E.; Yang, G.; Cancino, G.I. Consequences of Viral Infection and Cytokine Production During Pregnancy on Brain Development in Offspring. Front. Immunol. 2022, 13, 816619. [Google Scholar] [CrossRef] [PubMed]
- De Graaf-Peters, V.B.; Hadders-Algra, M. Ontogeny of the human central nervous system: What is happening when? Early Hum. Dev. 2006, 82, 257–266. [Google Scholar] [CrossRef]
- Boin, F.; Zanardini, R.; Pioli, R.; Altamura, C.; Maes, M.; Gennarelli, M. Association between -G308A tumor necrosis factor alpha gene polymorphism and schizophrenia. Mol. Psychiatry 2001, 6, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Presumey, J.; Bialas, A.R.; Carroll, M.C. Complement System in Neural Synapse Elimination in Development and Disease. Adv. Immunol. 2017, 135, 53–79. [Google Scholar] [PubMed]
- Yang, A.C.; Tsai, S.J. New Targets for Schizophrenia Treatment beyond the Dopamine Hypothesis. Int. J. Mol. Sci. 2017, 18, 1689. [Google Scholar] [CrossRef]
- Ganguli, S.; Chavali, P.L. Intrauterine Viral Infections: Impact of Inflammation on Fetal Neurodevelopment. Front. Neurosci. 2021, 15, 771557. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Symbol | Full Name | Biological Functions | Alteration in Gene Regulation in Schizophrenia | Related Study |
|---|---|---|---|---|
| APOL2 | Apolipoprotein L2 | Transport of lipids and binding of lipids to organelles. | The APOL 2 and 4 genes, located on chromosome 22q12.3–q13.1, are known to be upregulated in the brains of schizophrenic patients. | Lee et al., 2013 [121] |
| APOL4 | Apolipoprotein L4 | Exchange and transport of lipids/cholesterol. | Lee et al., 2013 [121] | |
| CHI3L1 | Chitinase 3 like 1 | Glycosyl hydrolase is associated with inflammation and tissue remodeling. | Gene expression studies indicate that CHI3L1 mRNA levels are increased in the hippocampus and dorsolateral prefrontal cortex of postmortem samples with schizophrenia compared with control samples. | Yang et al., 2008 [122] |
| MTHFR | Methylenetetrahydrofolate reductase | Conversion of 5,10-methylenetetrahydrofolate to 5-methyltetrahydrofolate. | MTHFR contributes to the regulation of RELN,56 whose expression is decreased in the hippocampus of subjects suffering from schizophrenia and major depressive disorder. It is possible that the low activity of MTHFR combined with other genetic and environmental factors contributes to the developmental outcome. | Favre et al., 2012 [123] and Kezurer et al., 2013 [124] |
| SETD1A | SET domain containing 1A, histone lysine methyltransferase | Histone methyltransferase complex. | Loss-of-function (LOF) variants that lead to disruption of SERD1A are likely to contribute to the etiology of neuropsychiatric disorders. | Takata et al., 2014 [125] |
| SYN2 | Synapsin II | Neuronal phosphoproteins associated with synaptogenesis and neurotransmitter release. | Decreased levels of synapsin II have been reported in schizophrenia. | Egbujo et al., 2015 [126] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kotsiri, I.; Resta, P.; Spyrantis, A.; Panotopoulos, C.; Chaniotis, D.; Beloukas, A.; Magiorkinis, E. Viral Infections and Schizophrenia: A Comprehensive Review. Viruses 2023, 15, 1345. https://doi.org/10.3390/v15061345
Kotsiri I, Resta P, Spyrantis A, Panotopoulos C, Chaniotis D, Beloukas A, Magiorkinis E. Viral Infections and Schizophrenia: A Comprehensive Review. Viruses. 2023; 15(6):1345. https://doi.org/10.3390/v15061345
Chicago/Turabian StyleKotsiri, Ioanna, Panagiota Resta, Alexandros Spyrantis, Charalampos Panotopoulos, Dimitrios Chaniotis, Apostolos Beloukas, and Emmanouil Magiorkinis. 2023. "Viral Infections and Schizophrenia: A Comprehensive Review" Viruses 15, no. 6: 1345. https://doi.org/10.3390/v15061345
APA StyleKotsiri, I., Resta, P., Spyrantis, A., Panotopoulos, C., Chaniotis, D., Beloukas, A., & Magiorkinis, E. (2023). Viral Infections and Schizophrenia: A Comprehensive Review. Viruses, 15(6), 1345. https://doi.org/10.3390/v15061345