

Roles and Mechanisms of NLRP3 in Influenza Viral Infection



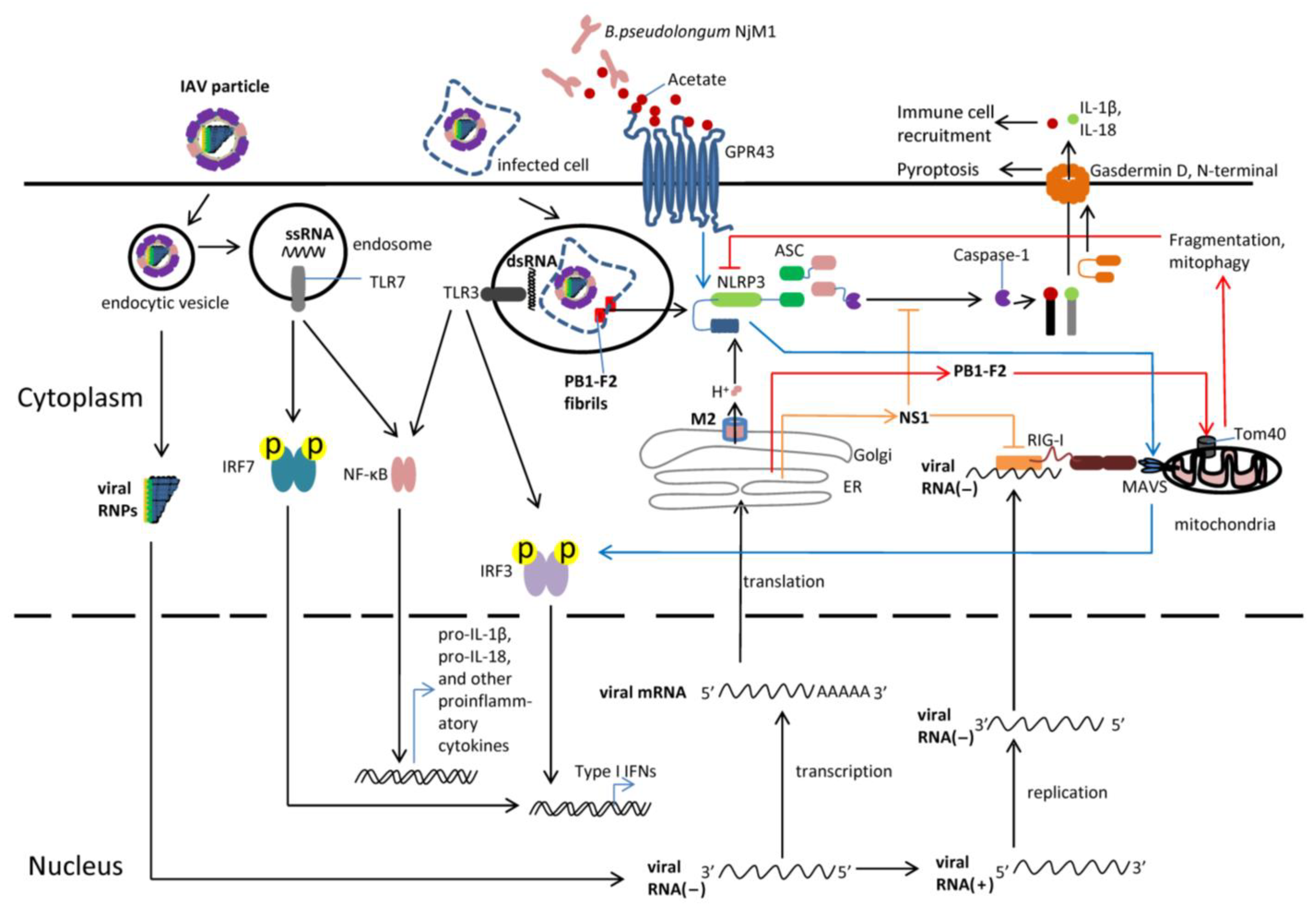{kind=link}
Abstract
1. Introduction
2. Influenza Virus
2.1. Characteristics of the Influenza Virus
2.2. Mutations of Influenza A Virus
3. Recognition of Influenza Virus by PRRs
3.1. Recognition of the IAV in the Cytoplasm
3.2. Recognition of the IAV in the Endosome and Phagosome
3.3. Evasion of the IAV from Immune Responses
4. Functions of NLRP3 Inflammasome in Influenza Virus Infection
4.1. Inflammasomes
4.2. NLRP3 Inflammasome during Influenza Virus Infection
4.2.1. The Impact of NLRP3 on Host Survival and Virus Clearance
4.2.2. Effect of NLRP3 on Cytokine Production and Cell Infiltration
4.2.3. NLRP3 and Lung Injury Repair
4.2.4. The Role of IL-18 and IL-1β in Influenza Virus Infection
4.2.5. The Role of Pyroptosis in Influenza Virus Infection
4.2.6. Additional Molecules Associated with NLRP3 Inflammasome in Influenza Virus Infection
4.2.7. Transition of NLRP3 from Detrimental to Protective Functions during Influenza Virus Infection
5. The Role of Commensal Microbiota in Influenza Virus Infection
6. Interplay between the NLRP3 Inflammasome and Microbiota
7. Perspectives for Future Research
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- WHO. Influenza (Seasonal) Fact Sheet. Available online: http://www.who.int/mediacentre/factsheets/fs211/en/ (accessed on 12 January 2023).
- Iuliano, A.D.; Roguski, K.M.; Chang, H.H.; Muscatello, D.J.; Palekar, R.; Tempia, S.; Cohen, C.; Gran, J.M.; Schanzer, D.; Cowling, B.J.; et al. Estimates of global seasonal influenza-associated respiratory mortality: A modelling study. Lancet 2018, 391, 1285–1300. [Google Scholar] [CrossRef] [PubMed]
- Taubenberger, J.K.; Morens, D.M. Influenza: The once and future pandemic. Public Health Rep. 2010, 125 (Suppl. 3), 16–26. [Google Scholar] [CrossRef]
- Eccles, R. Understanding the symptoms of the common cold and influenza. Lancet Infect. Dis. 2005, 5, 718–725. [Google Scholar] [CrossRef]
- Chertow, D.S.; Memoli, M.J. Bacterial coinfection in influenza: A grand rounds review. JAMA 2013, 309, 275–282. [Google Scholar] [CrossRef]
- Short, K.R.; Kroeze, E.; Fouchier, R.A.M.; Kuiken, T. Pathogenesis of influenza-induced acute respiratory distress syndrome. Lancet Infect. Dis. 2014, 14, 57–69. [Google Scholar] [CrossRef]
- Morens, D.M.; Taubenberger, J.K.; Fauci, A.S. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: Implications for pandemic influenza preparedness. J. Infect. Dis. 2008, 198, 962–970. [Google Scholar] [CrossRef]
- Bosch, A.A.; Biesbroek, G.; Trzcinski, K.; Sanders, E.A.; Bogaert, D. Viral and bacterial interactions in the upper respiratory tract. PLoS Pathog. 2013, 9, e1003057. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.L.; Pattemore, P.K.; Sanderson, G.; Smith, S.; Lampe, F.; Josephs, L.; Symington, P.; O’Toole, S.; Myint, S.H.; Tyrrell, D.A.; et al. Community study of role of viral infections in exacerbations of asthma in 9–11 year old children. BMJ 1995, 310, 1225–1229. [Google Scholar] [CrossRef]
- Sethi, S.; Murphy, T.F. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N. Engl. J. Med. 2008, 359, 2355–2365. [Google Scholar] [CrossRef]
- Kwong, J.C.; Schwartz, K.L.; Campitelli, M.A. Acute Myocardial Infarction after Laboratory-Confirmed Influenza Infection. N. Engl. J. Med. 2018, 378, 2540–2541. [Google Scholar] [CrossRef] [PubMed]
- Thompson, W.W.; Shay, D.K.; Weintraub, E.; Brammer, I.; Bridges, C.B.; Cox, N.J.; Fukuda, K. Influenza-associated hospitalizations in the United States. JAMA 2004, 292, 1333–1340. [Google Scholar] [CrossRef] [PubMed]
- O’Halloran, A.C.; Holstein, R.; Cummings, C.; Kirley, P.D.; Alden, N.B.; Yousey-Hindes, K.; Anderson, E.J.; Ryan, P.; Kim, S.; Lynfield, R.; et al. Rates of Influenza-Associated Hospitalization, Intensive Care Unit Admission, and In-Hospital Death by Race and Ethnicity in the United States From 2009 to 2019. JAMA Netw. Open 2021, 4, e2121880. [Google Scholar] [CrossRef]
- Niu, J.; Wu, S.; Chen, M.; Xu, K.; Guo, Q.; Lu, A.; Zhao, L.; Sun, B.; Meng, G. Hyperactivation of the NLRP3 inflammasome protects mice against influenza A virus infection via IL-1beta mediated neutrophil recruitment. Cytokine 2019, 120, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Allen, I.C.; Scull, M.A.; Moore, C.B.; Holl, E.K.; McElvania-TeKippe, E.; Taxman, D.J.; Guthrie, E.H.; Pickles, R.J.; Ting, J.P. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 2009, 30, 556–565. [Google Scholar] [CrossRef]
- Thomas, P.G.; Dash, P.; Aldridge, J.R., Jr.; Ellebedy, A.H.; Reynolds, C.; Funk, A.J.; Martin, W.J.; Lamkanfi, M.; Webby, R.J.; Boyd, K.L.; et al. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity 2009, 30, 566–575. [Google Scholar] [CrossRef]
- Iwasaki, A.; Pillai, P.S. Innate immunity to influenza virus infection. Nat. Rev. Immunol. 2014, 14, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Ichinohe, T.; Pang, I.K.; Kumamoto, Y.; Peaper, D.R.; Ho, J.H.; Murray, T.S.; Iwasaki, A. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc. Natl. Acad. Sci. USA 2011, 108, 5354–5359. [Google Scholar] [CrossRef]
- Chen, C.J.; Wu, G.H.; Kuo, R.L.; Shih, S.R. Role of the intestinal microbiota in the immunomodulation of influenza virus infection. Microbes Infect. 2017, 19, 570–579. [Google Scholar] [CrossRef]
- Wu, S.; Jiang, Z.Y.; Sun, Y.F.; Yu, B.; Chen, J.; Dai, C.Q.; Wu, X.L.; Tang, X.L.; Chen, X.Y. Microbiota regulates the TLR7 signaling pathway against respiratory tract influenza A virus infection. Curr. Microbiol. 2013, 67, 414–422. [Google Scholar] [CrossRef]
- Pielak, R.M.; Chou, J.J. Influenza M2 proton channels. Biochim. Biophys. Acta 2011, 1808, 522–529. [Google Scholar] [CrossRef]
- Subbarao, K.; Joseph, T. Scientific barriers to developing vaccines against avian influenza viruses. Nat. Rev. Immunol. 2007, 7, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Wiley, D.C.; Skehel, J.J. The structure and function of the hemagglutinin membrane glycoprotein of influenza virus. Annu. Rev. Biochem. 1987, 56, 365–394. [Google Scholar] [CrossRef]
- Skehel, J.J.; Wiley, D.C. Receptor binding and membrane fusion in virus entry: The influenza hemagglutinin. Annu. Rev. Biochem. 2000, 69, 531–569. [Google Scholar] [CrossRef] [PubMed]
- Palese, P.; Compans, R.W. Inhibition of influenza virus replication in tissue culture by 2-deoxy-2,3-dehydro-N-trifluoroacetylneuraminic acid (FANA): Mechanism of action. J. Gen. Virol. 1976, 33, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Pinto, L.H.; Lamb, R.A. The M2 proton channels of influenza A and B viruses. J. Biol. Chem. 2006, 281, 8997–9000. [Google Scholar] [CrossRef]
- Wang, J.; Li, F.; Ma, C. Recent progress in designing inhibitors that target the drug-resistant M2 proton channels from the influenza A viruses. Biopolymers 2015, 104, 291–309. [Google Scholar] [CrossRef] [PubMed]
- Portela, A.; Digard, P. The influenza virus nucleoprotein: A multifunctional RNA-binding protein pivotal to virus replication. J. Gen. Virol. 2002, 83, 723–734. [Google Scholar] [CrossRef]
- Ruigrok, R.W.; Barge, A.; Durrer, P.; Brunner, J.; Ma, K.; Whittaker, G.R. Membrane interaction of influenza virus M1 protein. Virology 2000, 267, 289–298. [Google Scholar] [CrossRef]
- Te Velthuis, A.J.W.; Fodor, E. Influenza virus RNA polymerase: Insights into the mechanisms of viral RNA synthesis. Nat. Rev. Microbiol. 2016, 14, 479–493. [Google Scholar] [CrossRef]
- Stasakova, J.; Ferko, B.; Kittel, C.; Sereinig, S.; Romanova, J.; Katinger, H.; Egorov, A. Influenza A mutant viruses with altered NS1 protein function provoke caspase-1 activation in primary human macrophages, resulting in fast apoptosis and release of high levels of interleukins 1beta and 18. J. Gen. Virol. 2005, 86, 185–195. [Google Scholar] [CrossRef]
- Diebold, S.S.; Montoya, M.; Unger, H.; Alexopoulou, L.; Roy, P.; Haswell, L.E.; Al-Shamkhani, A.; Flavell, R.; Borrow, P.; Sousa, C.R.E. Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature 2003, 424, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Naslund, T.I.; Liljestrom, P.; Weber, F.; Sousa, C.R.E. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 2006, 314, 997–1001. [Google Scholar] [CrossRef]
- Opitz, B.; Rejaibi, A.; Dauber, B.; Eckhard, J.; Vinzing, M.; Schmeck, B.; Hippenstiel, S.; Suttorp, N.; Wolff, T. IFN beta induction by influenza A virus is mediated by RIG-I which is regulated by the viral NS1 protein. Cell. Microbiol. 2007, 9, 930–938. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.; Zhu, X.; Li, Y.; Shi, M.; Zhang, J.; Bourgeois, M.; Yang, H.; Chen, X.; Recuenco, S.; Gomez, J.; et al. New world bats harbor diverse influenza A viruses. PLoS Pathog. 2013, 9, e1003657. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.; Li, Y.; Rivailler, P.; Conrardy, C.; Castillo, D.A.; Chen, L.M.; Recuenco, S.; Ellison, J.A.; Davis, C.T.; York, I.A.; et al. A distinct lineage of influenza A virus from bats. Proc. Natl. Acad. Sci. USA 2012, 109, 4269–4274. [Google Scholar] [CrossRef]
- Wille, M.; Holmes, E.C. The Ecology and Evolution of Influenza Viruses. Cold Spring Harb. Perspect. Med. 2020, 10, a038489. [Google Scholar] [CrossRef]
- Webster, R.G.; Laver, W.G.; Air, G.M.; Schild, G.C. Molecular mechanisms of variation in influenza viruses. Nature 1982, 296, 115–121. [Google Scholar] [CrossRef]
- Caton, A.J.; Brownlee, G.G.; Yewdell, J.W.; Gerhard, W. The antigenic structure of the influenza virus A/PR/8/34 hemagglutinin (H1 subtype). Cell 1982, 31, 417–427. [Google Scholar] [CrossRef]
- Scholtissek, C.; Rohde, W.; Von Hoyningen, V.; Rott, R. On the origin of the human influenza virus subtypes H2N2 and H3N2. Virology 1978, 87, 13–20. [Google Scholar] [CrossRef]
- Horimoto, T.; Kawaoka, Y. Pandemic Threat Posed by Avian Influenza A Viruses. Clin. Microbiol. Rev. 2001, 14, 129–149. [Google Scholar] [CrossRef]
- Tscherne, D.M.; García-Sastre, A. Virulence determinants of pandemic influenza viruses. J. Clin. Investig. 2011, 121, 6–13. [Google Scholar] [CrossRef]
- Gotch, F.; McMichael, A.; Smith, G.; Moss, B. Identification of viral molecules recognized by influenza-specific human cytotoxic T lymphocytes. J. Exp. Med. 1987, 165, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Voeten, J.T.; Bestebroer, T.M.; Nieuwkoop, N.J.; Fouchier, R.A.; Osterhaus, A.D.; Rimmelzwaan, G.F. Antigenic drift in the influenza A virus (H3N2) nucleoprotein and escape from recognition by cytotoxic T lymphocytes. J. Virol. 2000, 74, 6800–6807. [Google Scholar] [CrossRef]
- Seth, R.B.; Sun, L.; Chen, Z.J. Antiviral innate immunity pathways. Cell Res. 2006, 16, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Diebold, S.S.; Kaisho, T.; Hemmi, H.; Akira, S.; Reis e Sousa, C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 2004, 303, 1529–1531. [Google Scholar] [CrossRef] [PubMed]
- Le Goffic, R.; Balloy, V.; Lagranderie, M.; Alexopoulou, L.; Escriou, N.; Flavell, R.; Chignard, M.; Si-Tahar, M. Detrimental contribution of the Toll-like receptor (TLR)3 to influenza A virus-induced acute pneumonia. PLoS Pathog. 2006, 2, e53. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef]
- Ichinohe, T.; Lee, H.K.; Ogura, Y.; Flavell, R.; Iwasaki, A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J. Exp. Med. 2009, 206, 79–87. [Google Scholar] [CrossRef]
- Creager, H.M.; Kumar, A.; Zeng, H.; Maines, T.R.; Tumpey, T.M.; Belser, J.A. Infection and Replication of Influenza Virus at the Ocular Surface. J. Virol. 2018, 92, e02192-17. [Google Scholar] [CrossRef]
- Trompette, A.; Gollwitzer, E.S.; Pattaroni, C.; Lopez-Mejia, I.C.; Riva, E.; Pernot, J.; Ubags, N.; Fajas, L.; Nicod, L.P.; Marsland, B.J. Dietary Fiber Confers Protection against Flu by Shaping Ly6c− Patrolling Monocyte Hematopoiesis and CD8+ T Cell Metabolism. Immunity 2018, 48, 992–1005.e08. [Google Scholar] [CrossRef]
- Pothlichet, J.; Meunier, I.; Davis, B.K.; Ting, J.P.; Skamene, E.; von Messling, V.; Vidal, S.M. Type I IFN triggers RIG-I/TLR3/NLRP3-dependent inflammasome activation in influenza A virus infected cells. PLoS Pathog. 2013, 9, e1003256. [Google Scholar] [CrossRef] [PubMed]
- Van Der Sluijs, K.F.; Van Elden, L.J.; Arens, R.; Nijhuis, M.; Schuurman, R.; Florquin, S.; Kwakkel, J.; Akira, S.; Jansen, H.M.; Lutter, R.; et al. Enhanced viral clearance in interleukin-18 gene-deficient mice after pulmonary infection with influenza A virus. Immunology 2005, 114, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Pandey, K.P.; Zhou, Y. Influenza A Virus Infection Activates NLRP3 Inflammasome through Trans-Golgi Network Dispersion. Viruses 2022, 14, 88. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chen, Z.J. PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature 2018, 564, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhu, Y.; Lin, X.; Ren, C.; Zhao, J.; Wang, F.; Gao, X.; Xiao, R.; Zhao, L.; Chen, H.; et al. Influenza M2 protein regulates MAVS-mediated signaling pathway through interacting with MAVS and increasing ROS production. Autophagy 2019, 15, 1163–1181. [Google Scholar] [CrossRef]
- Wang, X.; Jiang, W.; Yan, Y.; Gong, T.; Han, J.; Tian, Z.; Zhou, R. RNA viruses promote activation of the NLRP3 inflammasome through a RIP1-RIP3-DRP1 signaling pathway. Nat. Immunol. 2014, 15, 1126–1133. [Google Scholar] [CrossRef]
- Le Goffic, R.; Pothlichet, J.; Vitour, D.; Fujita, T.; Meurs, E.; Chignard, M.; Si-Tahar, M. Cutting Edge: Influenza A virus activates TLR3-dependent inflammatory and RIG-I-dependent antiviral responses in human lung epithelial cells. J. Immunol. 2007, 178, 3368–3372. [Google Scholar] [CrossRef]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-kappa B by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef]
- McAuley, J.L.; Tate, M.D.; MacKenzie-Kludas, C.J.; Pinar, A.; Zeng, W.; Stutz, A.; Latz, E.; Brown, L.E.; Mansell, A. Activation of the NLRP3 inflammasome by IAV virulence protein PB1-F2 contributes to severe pathophysiology and disease. PLoS Pathog. 2013, 9, e1003392. [Google Scholar] [CrossRef]
- Yoshizumi, T.; Ichinohe, T.; Sasaki, O.; Otera, H.; Kawabata, S.; Mihara, K.; Koshiba, T. Influenza A virus protein PB1-F2 translocates into mitochondria via Tom40 channels and impairs innate immunity. Nat. Commun. 2014, 5, 4713. [Google Scholar] [CrossRef]
- Wang, R.; Zhu, Y.; Ren, C.; Yang, S.; Tian, S.; Chen, H.; Jin, M.; Zhou, H. Influenza A virus protein PB1-F2 impairs innate immunity by inducing mitophagy. Autophagy 2021, 17, 496–511. [Google Scholar] [CrossRef] [PubMed]
- Friesenhagen, J.; Viemann, D.; Borgeling, Y.; Schmolke, M.; Spiekermann, C.; Kirschnek, S.; Ludwig, S.; Roth, J. Highly pathogenic influenza viruses inhibit inflammatory response in monocytes via activation of rar-related orphan receptor RORalpha. J. Innate Immun. 2013, 5, 505–518. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Man, S.M.; Karki, R.; Kanneganti, T.D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 2017, 277, 61–75. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Broderick, L.; De Nardo, D.; Franklin, B.S.; Hoffman, H.M.; Latz, E. The Inflammasome and Autoinflammatory Syndromes. Annu. Rev. Pathol. 2014, 10, 395–424. [Google Scholar] [CrossRef]
- Xu, H.; Yang, J.; Gao, W.; Li, L.; Li, P.; Zhang, L.; Gong, Y.N.; Peng, X.; Xi, J.J.; Chen, S.; et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 2014, 513, 237–241. [Google Scholar] [CrossRef]
- Heneka, M.T.; McManus, R.M.; Latz, E. Inflammasome signalling in brain function and neurodegenerative disease. Nat. Rev. Neurosci. 2018, 19, 610–621. [Google Scholar] [CrossRef]
- Mayor, A.; Martinon, F.; De Smedt, T.; Petrilli, V.; Tschopp, J. A crucial function of SGT1 and HSP90 in inflammasome activity links mammalian and plant innate immune responses. Nat. Immunol. 2007, 8, 497–503. [Google Scholar] [CrossRef]
- Fernandes-Alnemri, T.; Wu, J.; Yu, J.W.; Datta, P.; Miller, B.; Jankowski, W.; Rosenberg, S.; Zhang, J.; Alnemri, E.S. The pyroptosome: A supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 2007, 14, 1590–1604. [Google Scholar] [CrossRef]
- Lu, A.; Magupalli, V.G.; Ruan, J.; Yin, Q.; Atianand, M.K.; Vos, M.R.; Schroder, G.F.; Fitzgerald, K.A.; Wu, H.; Egelman, E.H. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 2014, 156, 1193–1206. [Google Scholar] [CrossRef] [PubMed]
- Srinivasula, S.M.; Poyet, J.L.; Razmara, M.; Datta, P.; Zhang, Z.J.; Alnemri, E.S. The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J. Biol. Chem. 2002, 277, 21119–21122. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.; Ruegg, A.; Werner, S.; Beer, H.D. Active caspase-1 is a regulator of unconventional protein secretion. Cell 2008, 132, 818–831. [Google Scholar] [CrossRef]
- He, Y.; Hara, H.; Nunez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, H.; Shen, J.; Deng, R.; Yao, X.; Guo, Q.; Lu, A.; Sun, B.; Zhang, Y.; Meng, G. Gasdermin D Drives the Nonexosomal Secretion of Galectin-3, an Insulin Signal Antagonist. J. Immunol. 2019, 203, 2712–2723. [Google Scholar] [CrossRef]
- Stout-Delgado, H.W.; Vaughan, S.E.; Shirali, A.C.; Jaramillo, R.J.; Harrod, K.S. Impaired NLRP3 inflammasome function in elderly mice during influenza infection is rescued by treatment with nigericin. J. Immunol. 2012, 188, 2815–2824. [Google Scholar] [CrossRef]
- Shinya, K.; Gao, Y.; Cilloniz, C.; Suzuki, Y.; Fujie, M.; Deng, G.; Zhu, Q.; Fan, S.; Makino, A.; Muramoto, Y.; et al. Integrated clinical, pathologic, virologic, and transcriptomic analysis of H5N1 influenza virus-induced viral pneumonia in the rhesus macaque. J. Virol. 2012, 86, 6055–6066. [Google Scholar] [CrossRef] [PubMed]
- Tate, M.D.; Ong, J.D.H.; Dowling, J.K.; McAuley, J.L.; Robertson, A.B.; Latz, E.; Drummond, G.R.; Cooper, M.A.; Hertzog, P.J.; Mansell, A. Reassessing the role of the NLRP3 inflammasome during pathogenic influenza A virus infection via temporal inhibition. Sci. Rep. 2016, 6, 27912. [Google Scholar] [CrossRef]
- Ren, R.; Wu, S.; Cai, J.; Yang, Y.; Ren, X.; Feng, Y.; Chen, L.; Qin, B.; Xu, C.; Yang, H.; et al. The H7N9 influenza A virus infection results in lethal inflammation in the mammalian host via the NLRP3-caspase-1 inflammasome. Sci. Rep. 2017, 7, 7625. [Google Scholar] [CrossRef] [PubMed]
- Bruchard, M.; Rebe, C.; Derangere, V.; Togbe, D.; Ryffel, B.; Boidot, R.; Humblin, E.; Hamman, A.; Chalmin, F.; Berger, H.; et al. The receptor NLRP3 is a transcriptional regulator of TH2 differentiation. Nat. Immunol. 2015, 16, 859–870. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Gu, Y.; Zeng, X.; Wang, J. NLRP3 inflammasome regulates Th17 differentiation in rheumatoid arthritis. Clin. Immunol. 2018, 197, 154–160. [Google Scholar] [CrossRef]
- Park, S.H.; Ham, S.; Lee, A.; Moller, A.; Kim, T.S. NLRP3 negatively regulates Treg differentiation through Kpna2-mediated nuclear translocation. J. Biol. Chem. 2019, 294, 17951–17961. [Google Scholar] [CrossRef]
- Niu, J.; Cui, M.; Yang, X.; Li, J.; Yao, Y.; Guo, Q.; Lu, A.; Qi, X.; Zhou, D.; Zhang, C.; et al. Microbiota-derived acetate enhances host antiviral response via NLRP3. Nat. Commun. 2023, 14, 642. [Google Scholar] [CrossRef]
- Morales-Nebreda, L.; Chi, M.; Lecuona, E.; Chandel, N.S.; Dada, L.A.; Ridge, K.; Soberanes, S.; Nigdelioglu, R.; Sznajder, J.I.; Mutlu, G.M.; et al. Intratracheal administration of influenza virus is superior to intranasal administration as a model of acute lung injury. J. Virol. Methods 2014, 209, 116–120. [Google Scholar] [CrossRef]
- Liu, B. Interleukin-18 improves the early defence system against influenza virus infection by augmenting natural killer cell-mediated cytotoxicity. J. Gen. Virol. 2004, 85, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, N.; Kurrer, M.; Bachmann, M.F.; Kopf, M. Interleukin-1 is responsible for acute lung immunopathology but increases survival of respiratory influenza virus infection. J. Virol. 2005, 79, 6441–6448. [Google Scholar] [CrossRef]
- Huang, C.H.; Chen, C.J.; Yen, C.T.; Yu, C.P.; Huang, P.N.; Kuo, R.L.; Lin, S.J.; Chang, C.K.; Shih, S.R. Caspase-1 deficient mice are more susceptible to influenza A virus infection with PA variation. J. Infect. Dis. 2013, 208, 1898–1905. [Google Scholar] [CrossRef] [PubMed]
- Van den Eeckhout, B.; Tavernier, J.; Gerlo, S. Interleukin-1 as Innate Mediator of T Cell Immunity. Front. Immunol. 2021, 11, 621931. [Google Scholar] [CrossRef] [PubMed]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef]
- Kuriakose, T.; Kanneganti, T.D. Pyroptosis in Antiviral Immunity. In Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2019. [Google Scholar]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Owen, D.M.; Gale, M., Jr. Fighting the flu with inflammasome signaling. Immunity 2009, 30, 476–478. [Google Scholar] [CrossRef] [PubMed]
- Kuriakose, T.; Man, S.M.; Malireddi, R.K.S.; Karki, R.; Kesavardhana, S.; Place, D.E.; Neale, G.; Vogel, P.; Kanneganti, T.D. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci. Immunol. 2016, 1, aag2045. [Google Scholar] [CrossRef] [PubMed]
- Nogusa, S.; Thapa, R.J.; Dillon, C.P.; Liedmann, S.; Oguin, T.H., 3rd; Ingram, J.P.; Rodriguez, D.A.; Kosoff, R.; Sharma, S.; Sturm, O.; et al. RIPK3 Activates Parallel Pathways of MLKL-Driven Necroptosis and FADD-Mediated Apoptosis to Protect against Influenza A Virus. Cell Host Microbe 2016, 20, 13–24. [Google Scholar] [CrossRef]
- Thapa, R.J.; Ingram, J.P.; Ragan, K.B.; Nogusa, S.; Boyd, D.F.; Benitez, A.A.; Sridharan, H.; Kosoff, R.; Shubina, M.; Landsteiner, V.J.; et al. DAI Senses Influenza A Virus Genomic RNA and Activates RIPK3-Dependent Cell Death. Cell Host Microbe 2016, 20, 674–681. [Google Scholar] [CrossRef]
- Tate, M.D.; Mansell, A. An update on the NLRP3 inflammasome and influenza: The road to redemption or perdition? Curr. Opin. Immunol. 2018, 54, 80–85. [Google Scholar] [CrossRef]
- Jorgensen, I.; Rayamajhi, M.; Miao, E.A. Programmed cell death as a defence against infection. Nat. Rev. Immunol. 2017, 17, 151–164. [Google Scholar] [CrossRef]
- Kesavardhana, S.; Samir, P.; Zheng, M.; Malireddi, R.K.S.; Karki, R.; Sharma, B.R.; Place, D.E.; Briard, B.; Vogel, P.; Kanneganti, T.D. DDX3X coordinates host defense against influenza virus by activating the NLRP3 inflammasome and type I interferon response. J. Biol. Chem. 2021, 296, 100579. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, G.; Rogel, M.R.; Baker, M.A.; Troken, J.R.; Urich, D.; Morales-Nebreda, L.; Sennello, J.A.; Kutuzov, M.A.; Sitikov, A.; Davis, J.M.; et al. Vimentin regulates activation of the NLRP3 inflammasome. Nat. Commun. 2015, 6, 6574. [Google Scholar] [CrossRef]
- Nobre, C.C.; de Araujo, J.M.; Fernandes, T.A.; Cobucci, R.N.; Lanza, D.C.; Andrade, V.S.; Fernandes, J.V. Macrophage Migration Inhibitory Factor (MIF): Biological Activities and Relation with Cancer. Pathol. Oncol. Res. 2017, 23, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Ietta, F.; Ferro, E.A.V.; Bevilacqua, E.; Benincasa, L.; Maioli, E.; Paulesu, L. Role of the Macrophage Migration Inhibitory Factor (MIF) in the survival of first trimester human placenta under induced stress conditions. Sci. Rep. 2018, 8, 12150. [Google Scholar] [CrossRef]
- Lang, T.; Lee, J.P.W.; Elgass, K.; Pinar, A.A.; Tate, M.D.; Aitken, E.H.; Fan, H.P.; Creed, S.J.; Deen, N.S.; Traore, D.A.K.; et al. Macrophage migration inhibitory factor is required for NLRP3 inflammasome activation. Nat. Commun. 2018, 9, 2223. [Google Scholar] [CrossRef]
- Toldi, J.; Nemeth, D.; Hegyi, P.; Molnar, Z.; Solymar, M.; Farkas, N.; Alizadeh, H.; Rumbus, Z.; Pakai, E.; Garami, A. Macrophage migration inhibitory factor as a diagnostic and predictive biomarker in sepsis: Meta-analysis of clinical trials. Sci. Rep. 2021, 11, 8051. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.Z.; Chen, Q.Y.; Lan, H.Y. Macrophage Migration Inhibitory Factor (MIF) as a Stress Molecule in Renal Inflammation. Int. J. Mol. Sci. 2022, 23, 4908. [Google Scholar] [CrossRef]
- Xiang, Z.; Qu, F.; Li, J.; Qi, L.; Yang, Z.; Kong, X.; Yu, Z. Activator protein-1 (AP-1) and response to pathogen infection in the Hong Kong oyster (Crassostrea hongkongensis). Fish Shellfish. Immunol. 2014, 36, 83–89. [Google Scholar] [CrossRef]
- Papoudou-Bai, A.; Hatzimichael, E.; Barbouti, A.; Kanavaros, P. Expression patterns of the activator protein-1 (AP-1) family members in lymphoid neoplasms. Clin. Exp. Med. 2017, 17, 291–304. [Google Scholar] [CrossRef]
- Wan, P.; Zhang, S.M.; Ruan, Z.H.; Liu, X.L.; Yang, G.; Jia, Y.L.; Li, Y.K.; Pan, P.; Wang, W.B.; Li, G.; et al. AP-1 signaling pathway promotes pro-IL-1 beta transcription to facilitate NLRP3 inflammasome activation upon influenza A virus infection. Virulence 2022, 13, 502–513. [Google Scholar] [CrossRef]
- Gu, Y.; Zuo, X.; Zhang, S.; Ouyang, Z.; Jiang, S.; Wang, F.; Wang, G. The Mechanism behind Influenza Virus Cytokine Storm. Viruses 2021, 13, 1362. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Kanneganti, T.D. The regulation of the ZBP1-NLRP3 inflammasome and its implications in pyroptosis, apoptosis, and necroptosis (PANoptosis). Immunol. Rev. 2020, 297, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Goplen, N.P.; Sun, J. Aging and respiratory viral infection: From acute morbidity to chronic sequelae. Cell Biosci. 2021, 11, 112. [Google Scholar] [CrossRef]
- Manna, S.; Baindara, P.; Mandal, S.M. Molecular pathogenesis of secondary bacterial infection associated to viral infections including SARS-CoV-2. J. Infect. Public Health 2020, 13, 1397–1404. [Google Scholar] [CrossRef]
- Mishra, S.R.; Mahapatra, K.K.; Behera, B.P.; Patra, S.; Bhol, C.S.; Panigrahi, D.P.; Praharaj, P.P.; Singh, A.; Patil, S.; Dhiman, R.; et al. Mitochondrial dysfunction as a driver of NLRP3 inflammasome activation and its modulation through mitophagy for potential therapeutics. Int. J. Biochem. Cell Biol. 2021, 136, 106013. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; You, L.; Wu, J.; Zhao, M.; Guo, R.; Zhang, H.; Su, R.; Mao, Q.; Deng, D.; Hao, Y. Berberine suppresses influenza virus-triggered NLRP3 inflammasome activation in macrophages by inducing mitophagy and decreasing mitochondrial ROS. J. Leukoc. Biol. 2020, 108, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Sho, T.; Xu, J. Role and mechanism of ROS scavengers in alleviating NLRP3-mediated inflammation. Biotechnol. Appl. Biochem. 2019, 66, 4–13. [Google Scholar] [CrossRef]
- Moriyama, M.; Nagai, M.; Maruzuru, Y.; Koshiba, T.; Kawaguchi, Y.; Ichinohe, T. Influenza Virus-Induced Oxidized DNA Activates Inflammasomes. iScience 2020, 23, 101270. [Google Scholar] [CrossRef]
- Swiergiel, A.H.; Smagin, G.N.; Johnson, L.J.; Dunn, A.J. The role of cytokines in the behavioral responses to endotoxin and influenza virus infection in mice: Effects of acute and chronic administration of the interleukin-1-receptor antagonist (IL-1ra). Brain Res. 1997, 776, 96–104. [Google Scholar] [CrossRef]
- Wang, L.; Cai, J.; Zhao, X.; Ma, L.; Zeng, P.; Zhou, L.; Liu, Y.; Yang, S.; Cai, Z.; Zhang, S.; et al. Palmitoylation prevents sustained inflammation by limiting NLRP3 inflammasome activation through chaperone-mediated autophagy. Mol. Cell 2023, 83, 281–297.e10. [Google Scholar] [CrossRef]
- Jia, X.; Liu, B.; Bao, L.; Lv, Q.; Li, F.; Li, H.; An, Y.; Zhang, X.; Cao, B.; Wang, C. Delayed oseltamivir plus sirolimus treatment attenuates H1N1 virus-induced severe lung injury correlated with repressed NLRP3 inflammasome activation and inflammatory cell infiltration. PLoS Pathog. 2018, 14, e1007428. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.R.; Pop, M.; Deboy, R.T.; Eckburg, P.B.; Turnbaugh, P.J.; Samuel, B.S.; Gordon, J.I.; Relman, D.A.; Fraser-Liggett, C.M.; Nelson, K.E. Metagenomic analysis of the human distal gut microbiome. Science 2006, 312, 1355–1359. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L. The gut microbiota and obesity: From correlation to causality. Nat. Rev. Microbiol. 2013, 11, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L. Genomics: The tale of our other genome. Nature 2010, 465, 879–880. [Google Scholar] [CrossRef]
- Lederberg, J. Infectious history. Science 2000, 288, 287–293. [Google Scholar] [CrossRef]
- Backhed, F.; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. Host-bacterial mutualism in the human intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef]
- Andersson, A.F.; Lindberg, M.; Jakobsson, H.; Backhed, F.; Nyren, P.; Engstrand, L. Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS ONE 2008, 3, e2836. [Google Scholar] [CrossRef]
- Nam, Y.D.; Jung, M.J.; Roh, S.W.; Kim, M.S.; Bae, J.W. Comparative analysis of Korean human gut microbiota by barcoded pyrosequencing. PLoS ONE 2011, 6, e22109. [Google Scholar] [CrossRef]
- Capparelli, R.; Cuomo, P.; Gentile, A.; Iannelli, D. Microbiota-Liver Diseases Interactions. Int. J. Mol. Sci. 2023, 24, 3883. [Google Scholar] [CrossRef]
- Van Hul, M.; Cani, P.D. The gut microbiota in obesity and weight management: Microbes as friends or foe? Nat. Rev. Endocrinol. 2023, 19, 258–271. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, B.; Chen, H.; Yang, F.; Huang, J.L.; Jiao, X.A.; Zhang, Y.Z. Environmental factors and gut microbiota: Toward better conservation of deer species. Front. Microbiol. 2023, 14, 1136413. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.P.; Shao, J.J.; Cui, Q.H.; Ni, W.T.; Yang, Y.; Yan, B.B. Bioactivities of Dietary Polyphenols and Their Effects on Intestinal Microbiota. Mini-Rev. Med. Chem. 2023, 23, 361–377. [Google Scholar] [CrossRef] [PubMed]
- Zysset-Burri, D.C.; Morandi, S.; Herzog, E.L.; Berger, L.E.; Zinkernagel, M.S. The role of the gut microbiome in eye diseases. Prog. Retin. Eye Res. 2023, 92, 101117. [Google Scholar] [CrossRef]
- Donald, K.; Finlay, B.B. Early-life interactions between the microbiota and immune system: Impact on immune system development and atopic disease. Nat. Rev. Immunol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Lubin, J.B.; Green, J.; Maddux, S.; Denu, L.; Duranova, T.; Lanza, M.; Wynosky-Dolfi, M.; Flores, J.N.; Grimes, L.P.; Brodsky, I.E.; et al. Arresting microbiome development limits immune system maturation and resistance to infection in mice. Cell Host Microbe 2023, 31, 554–570.e7. [Google Scholar] [CrossRef]
- Lukacova, I.; Ambro, L.; Dubayova, K.; Marekova, M. The gut microbiota, its relationship to the immune system, and possibilities of its modulation. Epidemiol. Mikrobiol. Imunol. 2023, 72, 40–53. [Google Scholar]
- Wilks, J.; Golovkina, T. Influence of microbiota on viral infections. PLoS Pathog. 2012, 8, e1002681. [Google Scholar] [CrossRef]
- He, Y.; Wen, Q.; Yao, F.; Xu, D.; Huang, Y.; Wang, J. Gut-lung axis: The microbial contributions and clinical implications. Crit. Rev. Microbiol. 2017, 43, 81–95. [Google Scholar] [CrossRef]
- Budden, K.F.; Gellatly, S.L.; Wood, D.L.; Cooper, M.A.; Morrison, M.; Hugenholtz, P.; Hansbro, P.M. Emerging pathogenic links between microbiota and the gut-lung axis. Nat. Rev. Microbiol. 2017, 15, 55–63. [Google Scholar] [CrossRef]
- Abt, M.C.; Osborne, L.C.; Monticelli, L.A.; Doering, T.A.; Alenghat, T.; Sonnenberg, G.F.; Paley, M.A.; Antenus, M.; Williams, K.L.; Erikson, J.; et al. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity 2012, 37, 158–170. [Google Scholar] [CrossRef]
- Feng, J.; Gao, X.; Chen, X.; Tong, X.; Qian, M.; Gao, H.; Wang, J.; Wang, S.; Fei, C.; Cao, L.; et al. Mechanism of Jinzhen Oral Liquid against influenza-induced lung injury based on metabonomics and gut microbiome. J. Ethnopharmacol. 2023, 303, 115977. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Limaye, A.; Liu, J.R.; Wu, T.N. Potential probiotics for regulation of the gut-lung axis to prevent or alleviate influenza in vulnerable populations. J. Tradit. Complement. Med. 2023, 13, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Xie, R.; Zhang, H.; Zhang, H.; Li, C.; Cui, D.; Li, S.; Li, Z.; Liu, H.; Huang, J. Hemagglutinin expressed by yeast reshapes immune microenvironment and gut microbiota to trigger diverse anti-infection response in infected birds. Front. Immunol. 2023, 14, 1125190. [Google Scholar] [CrossRef]
- Deng, L.; Shi, Y.C.; Liu, P.; Wu, S.Z.; Lv, Y.W.; Xu, H.C.; Chen, X.Y. GeGen QinLian decoction alleviate influenza virus infectious pneumonia through intestinal flora. Biomed. Pharmacother. 2021, 141, 111896. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.Y.; Chen, X.L.; Zhou, L.; Li, J.Y.; Wang, D.; Yang, W.T.; Wu, H.; Yao, J.Y.; Yang, G.L.; Wang, C.F.; et al. The gut microbiota of bats confers tolerance to influenza virus (H1N1) infection in mice. Transbound. Emerg. Dis. 2022, 69, E1469–E1487. [Google Scholar] [CrossRef]
- Gao, J.; Chen, H.; Xu, L.; Li, S.; Yan, H.; Jiang, L.; Cheng, W.; Jiang, Z. Effects of Intestinal Microorganisms on Influenza-Infected Mice with Antibiotic-Induced Intestinal Dysbiosis, through the TLR7 Signaling Pathway. Front. Biosci. (Landmark Ed.) 2023, 28, 43. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.H.; Pomare, E.W.; Branch, W.J.; Naylor, C.P.; Macfarlane, G.T. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 1987, 28, 1221–1227. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, G.T.; Macfarlane, S. Bacteria, colonic fermentation, and gastrointestinal health. J. AOAC Int. 2012, 95, 50–60. [Google Scholar] [CrossRef]
- Rey, F.E.; Faith, J.J.; Bain, J.; Muehlbauer, M.J.; Stevens, R.D.; Newgard, C.B.; Gordon, J.I. Dissecting the in vivo metabolic potential of two human gut acetogens. J. Biol. Chem. 2010, 285, 22082–22090. [Google Scholar] [CrossRef]
- Louis, P.; Hold, G.L.; Flint, H.J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [Google Scholar] [CrossRef]
- Ragsdale, S.W.; Pierce, E. Acetogenesis and the Wood-Ljungdahl pathway of CO2 fixation. Biochim. Biophys. Acta 2008, 1784, 1873–1898. [Google Scholar] [CrossRef] [PubMed]
- Stefan, K.L.; Kim, M.V.; Iwasaki, A.; Kasper, D.L. Commensal Microbiota Modulation of Natural Resistance to Virus Infection. Cell 2020, 183, 1312–1324.e10. [Google Scholar] [CrossRef] [PubMed]
- Steed, A.L.; Christophi, G.P.; Kaiko, G.E.; Sun, L.; Goodwin, V.M.; Jain, U.; Esaulova, E.; Artyomov, M.N.; Morales, D.J.; Holtzman, M.J.; et al. The microbial metabolite desaminotyrosine protects from influenza through type I interferon. Science 2017, 357, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Schaupp, L.; Muth, S.; Rogell, L.; Kofoed-Branzk, M.; Melchior, F.; Lienenklaus, S.; Ganal-Vonarburg, S.C.; Klein, M.; Guendel, F.; Hain, T.; et al. Microbiota-Induced Type I Interferons Instruct a Poised Basal State of Dendritic Cells. Cell 2020, 181, 1080–1096.e19. [Google Scholar] [CrossRef]
- Akatsu, H.; Nagafuchi, S.; Kurihara, R.; Okuda, K.; Kanesaka, T.; Ogawa, N.; Kanematsu, T.; Takasugi, S.; Yamaji, T.; Takami, M.; et al. Enhanced vaccination effect against influenza by prebiotics in elderly patients receiving enteral nutrition. Geriatr. Gerontol. Int. 2016, 16, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Hagan, T.; Cortese, M.; Rouphael, N.; Boudreau, C.; Linde, C.; Maddur, M.S.; Das, J.; Wang, H.; Guthmiller, J.; Zheng, N.Y.; et al. Antibiotics-Driven Gut Microbiome Perturbation Alters Immunity to Vaccines in Humans. Cell 2019, 178, 1313–1328.e13. [Google Scholar] [CrossRef]
- Borey, M.; Blanc, F.; Lemonnier, G.; Leplat, J.J.; Jardet, D.; Rossignol, M.N.; Ravon, L.; Billon, Y.; Bernard, M.; Estelle, J.; et al. Links between fecal microbiota and the response to vaccination against influenza A virus in pigs. npj Vaccines 2021, 6, 92. [Google Scholar] [CrossRef]
- Goncalves, J.I.B.; Borges, T.J.; de Souza, A.P.D. Microbiota and the Response to Vaccines Against Respiratory Virus. Front. Immunol. 2022, 13, 889945. [Google Scholar] [CrossRef]
- Li, R.; Lim, A.; Ow, S.T.; Phoon, M.C.; Locht, C.; Chow, V.T.; Alonso, S. Development of live attenuated Bordetella pertussis strains expressing the universal influenza vaccine candidate M2e. Vaccine 2011, 29, 5502–5511. [Google Scholar] [CrossRef]
- Demento, S.L.; Cui, W.; Criscione, J.M.; Stern, E.; Tulipan, J.; Kaech, S.M.; Fahmy, T.M. Role of sustained antigen release from nanoparticle vaccines in shaping the T cell memory phenotype. Biomaterials 2012, 33, 4957–4964. [Google Scholar] [CrossRef]
- Reed, S.G.; Orr, M.T.; Fox, C.B. Key roles of adjuvants in modern vaccines. Nat. Med. 2013, 19, 1597–1608. [Google Scholar] [CrossRef] [PubMed]
- McKee, A.S.; Marrack, P. Old and new adjuvants. Curr. Opin. Immunol. 2017, 47, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Iwabuchi, N.; Xiao, J.Z.; Yaeshima, T.; Iwatsuki, K. Oral administration of Bifidobacterium longum ameliorates influenza virus infection in mice. Biol. Pharm. Bull. 2011, 34, 1352–1355. [Google Scholar] [CrossRef] [PubMed]
- Kawase, M.; He, F.; Kubota, A.; Harata, G.; Hiramatsu, M. Oral administration of lactobacilli from human intestinal tract protects mice against influenza virus infection. Lett. Appl. Microbiol. 2010, 51, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Waki, N.; Yajima, N.; Suganuma, H.; Buddle, B.M.; Luo, D.; Heiser, A.; Zheng, T. Oral administration of Lactobacillus brevis KB290 to mice alleviates clinical symptoms following influenza virus infection. Lett. Appl. Microbiol. 2014, 58, 87–93. [Google Scholar] [CrossRef]
- Goto, H.; Sagitani, A.; Ashida, N.; Kato, S.; Hirota, T.; Shinoda, T.; Yamamoto, N. Anti-influenza virus effects of both live and non-live Lactobacillus acidophilus L-92 accompanied by the activation of innate immunity. Br. J. Nutr. 2013, 110, 1810–1818. [Google Scholar] [CrossRef]
- Park, M.K.; Ngo, V.; Kwon, Y.M.; Lee, Y.T.; Yoo, S.; Cho, Y.H.; Hong, S.M.; Hwang, H.S.; Ko, E.J.; Jung, Y.J.; et al. Lactobacillus plantarum DK119 as a Probiotic Confers Protection against Influenza Virus by Modulating Innate Immunity. PLoS ONE 2013, 8, e75368. [Google Scholar] [CrossRef]
- Maeda, N.; Nakamura, R.; Hirose, Y.; Murosaki, S.; Yamamoto, Y.; Kase, T.; Yoshikai, Y. Oral administration of heat-killed Lactobacillus plantarum L-137 enhances protection against influenza virus infection by stimulation of type I interferon production in mice. Int. Immunopharmacol. 2009, 9, 1122–1125. [Google Scholar] [CrossRef]
- Kechaou, N.; Chain, F.; Gratadoux, J.J.; Blugeon, S.; Bertho, N.; Chevalier, C.; Le Goffic, R.; Courau, S.; Molimard, P.; Chatel, J.M.; et al. Identification of one novel candidate probiotic Lactobacillus plantarum strain active against influenza virus infection in mice by a large-scale screening. Appl. Environ. Microbiol. 2013, 79, 1491–1499. [Google Scholar] [CrossRef]
- Kikuchi, Y.; Kunitoh-Asari, A.; Hayakawa, K.; Imai, S.; Kasuya, K.; Abe, K.; Adachi, Y.; Fukudome, S.; Takahashi, Y.; Hachimura, S. Oral administration of Lactobacillus plantarum strain AYA enhances IgA secretion and provides survival protection against influenza virus infection in mice. PLoS ONE 2014, 9, e86416. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, L.; Zhang, J.; Huang, K.; Sun, X.; Yang, Y.; Wang, T.; Zhang, Q.; Zou, Z.; Jin, M. Oral or intranasal immunization with recombinant Lactobacillus plantarum displaying head domain of Swine Influenza A virus hemagglutinin protects mice from H1N1 virus. Microb. Cell Factories 2022, 21, 185. [Google Scholar] [CrossRef]
- Takahashi, C.; Kozawa, M. The effect of partially hydrolyzed guar gum on preventing influenza infection. Clin. Nutr. ESPEN 2021, 42, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Zhang, C.; Xing, Y.; Xue, G.; Zhang, Q.; Pan, F.; Wu, G.; Hu, Y.; Guo, Q.; Lu, A.; et al. Remodelling of the gut microbiota by hyperactive NLRP3 induces regulatory T cells to maintain homeostasis. Nat. Commun. 2017, 8, 1896. [Google Scholar] [CrossRef] [PubMed]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef]
- Macia, L.; Tan, J.; Vieira, A.T.; Leach, K.; Stanley, D.; Luong, S.; Maruya, M.; McKenzie, C.I.; Hijikata, A.; Wong, C.; et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat. Commun. 2015, 6, 6734. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.D.; Jiang, Z.Y.; Wang, C.L.; Li, N.; Bo, L.L.; Zha, Y.P.; Bian, J.J.; Zhang, Y.; Deng, X.M. Acetate attenuates inflammasome activation through GPR43-mediated Ca2+-dependent NLRP3 ubiquitination. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Franchi, L.; Munoz-Planillo, R.; Nunez, G. Sensing and reacting to microbes through the inflammasomes. Nat. Immunol. 2012, 13, 325–332. [Google Scholar] [CrossRef]
- Levy, M.; Thaiss, C.A.; Zeevi, D.; Dohnalova, L.; Zilberman-Schapira, G.; Mahdi, J.A.; David, E.; Savidor, A.; Korem, T.; Herzig, Y.; et al. Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell 2015, 163, 1428–1443. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niu, J.; Meng, G. Roles and Mechanisms of NLRP3 in Influenza Viral Infection. Viruses 2023, 15, 1339. https://doi.org/10.3390/v15061339
Niu J, Meng G. Roles and Mechanisms of NLRP3 in Influenza Viral Infection. Viruses. 2023; 15(6):1339. https://doi.org/10.3390/v15061339
Chicago/Turabian StyleNiu, Junling, and Guangxun Meng. 2023. "Roles and Mechanisms of NLRP3 in Influenza Viral Infection" Viruses 15, no. 6: 1339. https://doi.org/10.3390/v15061339
APA StyleNiu, J., & Meng, G. (2023). Roles and Mechanisms of NLRP3 in Influenza Viral Infection. Viruses, 15(6), 1339. https://doi.org/10.3390/v15061339