
Retrospective Spatio-Temporal Dynamics of Dengue Virus 1, 2 and 4 in Paraguay


 ,
,  , , , , , , , ,
, , , , , , , ,  , , , ,
, , , ,  ,
,  add
Show full author list
add
Show full author list
Abstract
1. Introduction
2. Materials and Methods
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization, WHO. Geographical Expansion of Cases of Dengue and Chikungunya Beyond the Historical Areas of Transmission in the Region of the Americas. 2023. Available online: https://www.who.int/emergencies/disease-outbreak-news/item/2023-DON448 (accessed on 15 April 2023).
- Gómez Gómez, R.E.; Kim, J.; Hong, K.; Jang, J.Y.; Kisiju, T.; Kim, S.; Chun, B.C. Association between Climate Factors and Dengue Fever in Asuncion, Paraguay: A Generalized Additive Model. Int. J. Environ. Res. Public Health 2022, 26, 12192. [Google Scholar] [CrossRef] [PubMed]
- Gräf, T.; Vazquez, C.; Giovanetti, M.; de Bruycker-Nogueira, F.; Fonseca, V.; Claro, I.M.; de Jesus, J.G.; Gómez, A.; Xavier, J.; de Mendonça, M.C.L.; et al. Epidemiologic History and Genetic Diversity Origins of Chikungunya and Dengue Viruses, Paraguay. Emerg. Infect. Dis. 2021, 27, 1393–1404. [Google Scholar] [CrossRef] [PubMed]
- Gardy, J.L.; Loman, N.J. Towards a genomics-informed, real-time, global pathogen surveillance system. Nat. Rev. Genet. 2018, 19, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Lanciotti, R.S.; Kosoy, O.L.; Laven, J.J.; Velez, J.O.; Lambert, A.J.; Johnson, A.J.; Stanfield, S.M.; Duffy, M.R. Genetic and serologic properties of Zika virus associated with an epidemic, Yap State, Micronesia, 2007. Emerg. Infect. Dis. 2008, 14, 1232–1239. [Google Scholar] [CrossRef]
- Santiago, G.A.; Vergne, E.; Quiles, Y.; Cosme, J.; Vazquez, J.; Medina, J.F.; Medina, F.; Colón, C.; Margolis, H.; Muñoz-Jordán, J.L. Analytical and clinical performance of the CDC real time RT-PCR assay for detection and typing of dengue virus. PLoS Negl. Trop. Dis. 2013, 7, e2311. [Google Scholar] [CrossRef]
- Lanciotti, R.S.; Kosoy, O.L.; Laven, J.J.; Panella, A.J.; Velez, J.O.; Lambert, A.J.; Campbell, G.L. Chikungunya virus in US travelers returning from India, 2006. Emerg. Infect. Dis. 2007, 13, 764–767. [Google Scholar] [CrossRef]
- Quick, J.; Grubaugh, N.D.; Pullan, S.T.; Claro, I.M.; Smith, A.D.; Gangavarapu, K.; Oliveira, G.; Robles-Sikisaka, R.; Rogers, T.F.; Beutler, N.A.; et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 2017, 12, 261–276. [Google Scholar] [CrossRef]
- Vilsker, M.; Moosa, Y.; Nooij, S.; Fonseca, V.; Ghysens, Y.; Dumon, K.; Pauwels, R.; Alcantara, L.C.; Vanden Eynden, E.; Vandamme, A.-M.; et al. Genome Detective: An automated system for virus identification from high-throughput sequencing data. Bioinformatics 2019, 1, 871–873. [Google Scholar] [CrossRef]
- Fonseca, V.; Libin, P.J.K.; Theys, K.; Faria, N.R.; Nunes, M.R.T.; Restovic, M.I.; Freire, M.; Giovanetti, M.; Cuypers, L.; Nowé, A.; et al. A computational method for the identification of dengue, Zika and chikungunya virus species and genotypes. PLoS Negl. Trop. Dis. 2019, 13, 29–30. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinformatics. Brief. Bioinform. 2019, 19, 1160–1166. [Google Scholar] [CrossRef]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 3, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 2, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Baele, G.; Li, W.L.; Drummond, A.J.; Suchard, M.A.; Lemey, P. Accurate model selection of relaxed molecular clocks in bayesian phylogenetics. Mol. Biol. Evol. 2013, 30, 239–243. [Google Scholar] [CrossRef]
- Lemey, P.; Rambaut, A.; Welch, J.J.; Suchard, M.A. Phylogeography takes a relaxed random walk in continuous space and time. Mol. Biol. Evol. 2010, 27, 1877–1885. [Google Scholar] [CrossRef]
- Pybus, O.G.; Suchard, M.A.; Lemey, P.; Bernardin, F.J.; Rambaut, A.; Crawford, F.W.; Gray, R.R.; Arinaminpathy, N.; Stramer, S.L.; Busch, M.P.; et al. Unifying the spatial epidemiology and molecular evolution of emerging epidemics. Proc. Natl. Acad. Sci. USA 2012, 109, 15066–15071. [Google Scholar] [CrossRef]
- Dellicour, S.; Gill, M.S.; Faria, N.R.; Rambaut, A.; Pybus, O.G.; A Suchard, M.; Lemey, P. Relax, keep walking—A practical guide to continuous phylogeographic inference with BEAST. Mol. Biol. Evol. 2021, 38, 3486–3493. [Google Scholar] [CrossRef]
- Dellicour, S.; Rose, R.; Faria, N.R.; Lemey, P.; Pybus, O.G. SERAPHIM:studying environmental rasters and phylogenetically informed movements. Bioinformatics 2016, 32, 3204–3206. [Google Scholar] [CrossRef]
- Pan-American Health Organization (PAHO). Dengue Report. 2023. Available online: https://www3.paho.org/data/index.php/en/mnu-topics/indicadores-dengue-en.html (accessed on 10 April 2023).
- Nakase, T.; Giovanetti, M.; Obolski, U.; Lourenço, J. Global transmission suitability maps for dengue virus transmitted by Aedes aegypti from 1981 to 2019. MedRxiv 2023. [Google Scholar] [CrossRef]
- Adelino, T.É.R.; Giovanetti, M.; Fonseca, V.; Xavier, J.; de Abreu, Á.S.; Nascimento, V.A.D.; Demarchi, L.H.F.; Oliveira, M.A.A.; da Silva, V.L.; de Mello, A.L.E.S.; et al. Field and classroom initiatives for portable sequence-based monitoring of dengue virus in Brazil. Nat. Commun. 2021, 16, 12–2296. [Google Scholar] [CrossRef] [PubMed]
- Ministerio de Salud Publica, Paraguay. 2023. Available online: https://dgvs.mspbs.gov.py/arbovirosis/ (accessed on 15 April 2023).



{kind=link}
{kind=link}
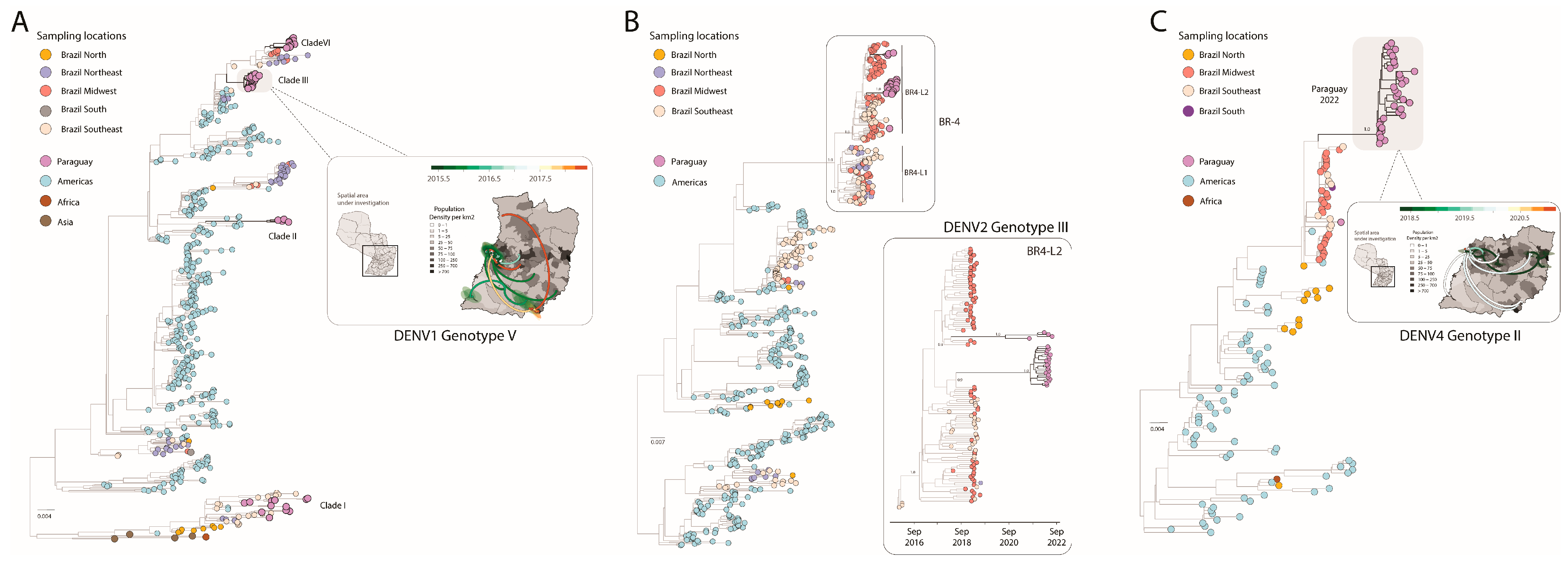
| Accession Number | Virus | Lineages | Country | Ct | City | Collection Date | Date Onset Symptoms | Gender | Age | Coverage |
|---|---|---|---|---|---|---|---|---|---|---|
| OQ567774 | DENV-1 | Genotype V | PY | 24 | Salto Del Guaira | 27-Feb-2014 | 27-Feb-2014 | M | 27 | 88.7 |
| OQ567775 | DENV-1 | Genotype V | PY | 22 | Santa Rosa Del Aguaray | 23-Mar-2015 | 20-Mar-2015 | M | 28 | 88.5 |
| OQ567776 | DENV-1 | Genotype V | PY | 20 | Santa Rosa Del Aguaray | 20-Mar-2015 | 18-Mar-2015 | F | 41 | 93.6 |
| OQ567777 | DENV-1 | Genotype V | PY | 20 | Santa Rosa Del Aguaray | 26-Mar-2015 | 25-Mar-2015 | F | 27 | 93.2 |
| OQ567778 | DENV-1 | Genotype V | PY | 24 | Carapegua | 13-May-2015 | 12-May-2015 | M | 43 | 89.3 |
| OQ567779 | DENV-1 | Genotype V | PY | 26 | Caazapa | 2-Jun-2015 | 31-May-2015 | M | 46 | 89.2 |
| OQ567780 | DENV-1 | Genotype V | PY | 18 | Ciudad del Este | 15-Dec-2015 | 14-Dec-2015 | M | 41 | 93 |
| OQ567781 | DENV-1 | Genotype V | PY | 19 | Asuncion | 18-Dec-2015 | 17-Dec-2015 | F | 22 | 93.4 |
| OQ567782 | DENV-1 | Genotype V | PY | 20 | Concepcion | 17-Dec-2015 | 14-Dec-2015 | F | 36 | 94.4 |
| OQ567783 | DENV-1 | Genotype V | PY | 22 | Concepcion | 23-Dec-2015 | 21-Dec-2015 | F | 53 | 88.8 |
| OQ567784 | DENV-1 | Genotype V | PY | 21 | Benjamin Aceval | 21-Dec-2015 | 20-Dec-2015 | M | SD | 88.8 |
| OQ567785 | DENV-1 | Genotype V | PY | 25 | San Juan Nepomuceno | 24-Dec-2015 | 22-Dec-2015 | F | 19 | 88.7 |
| OQ567786 | DENV-1 | Genotype V | PY | 27 | Itapua | 29-Jan-2016 | 26-Jan-2016 | M | 18 | 88.5 |
| OQ567787 | DENV-1 | Genotype V | PY | 22 | Pilar | 4-Feb-2016 | 2-Feb-2016 | F | 24 | 88.8 |
| OQ567788 | DENV-1 | Genotype V | PY | 25 | Pilar | 2-Apr-2016 | 2-Mar-2016 | F | 24 | 92.9 |
| OQ567789 | DENV-1 | Genotype V | PY | 26 | Yatytay | 2-Jan-2016 | 2-Jan-2016 | F | 83 | 92.9 |
| OQ567790 | DENV-1 | Genotype V | PY | 26 | San Lorenzo | 24-Jan-2016 | 23-Jan-2016 | F | 23 | 92.6 |
| OQ567791 | DENV-1 | Genotype V | PY | 28 | Asuncion | 10-Feb-2016 | 2-Jun-2016 | F | 28 | 89 |
| OQ567792 | DENV-1 | Genotype V | PY | 26 | Carapegua | 2-Sep-2016 | 2-Aug-2016 | M | 18 | 92.7 |
| OQ567793 | DENV-1 | Genotype V | PY | 21 | Pirayu | 2-Sep-2016 | 2-Jun-2016 | F | 40 | 94.1 |
| OQ567794 | DENV-1 | Genotype V | PY | 28 | Coronel Oviedo | 2-Oct-2016 | 2-Aug-2016 | F | 37 | 84.1 |
| OQ567795 | DENV-1 | Genotype V | PY | 22 | Itaugua | 14-Feb-2016 | 14-Feb-2016 | F | 23 | 93.1 |
| OQ567796 | DENV-1 | Genotype V | PY | 26 | Mariscal Estigarribia | 3-Jan-2017 | 28-Feb-2017 | F | 61 | 92.9 |
| OQ567797 | DENV-1 | Genotype V | PY | 27 | Asuncion | 29-Mar-2017 | 28-Mar-2017 | F | 29 | 89.2 |
| OQ567798 | DENV-1 | Genotype V | PY | 21 | Paraguari | 26-Apr-2017 | 25-Apr-2017 | F | 28 | 89.3 |
| OQ567799 | DENV-1 | Genotype V | PY | 21 | Tte. Irala Fernandez | 25-Apr-2017 | 23-Apr-2017 | F | 44 | 93.3 |
| OQ567800 | DENV-1 | Genotype V | PY | 24 | Filadeflia | 27-Mar-2017 | 26-Mar-2017 | M | 25 | 92.9 |
| OQ567801 | DENV-1 | Genotype V | PY | 27 | San Lorenzo | 29-Apr-2017 | 26-Apr-2017 | F | 25 | 88.9 |
| OQ567802 | DENV-1 | Genotype V | PY | 27 | Capitan Bado | 22-Mar-2022 | 21-Mar-2022 | F | 18 | 93 |
| OQ567803 | DENV-1 | Genotype V | PY | 22 | Capitan Bado | 28-Mar-2022 | 26-Mar-2022 | M | 18 | 89.6 |
| OQ567804 | DENV-1 | Genotype V | PY | 25 | Capitan Bado | 4-Apr-2022 | 1-Apr-2022 | M | 56 | 88.1 |
| OQ567805 | DENV-1 | Genotype V | PY | 26 | Guarambare | 25-Mar-2022 | 23-Mar-2022 | M | 41 | 94.6 |
| OQ567806 | DENV-1 | Genotype V | PY | 26 | Capitan Bado | 4-Dec-2022 | 11-Apr-2022 | F | 57 | 91.8 |
| OQ567807 | DENV-1 | Genotype V | PY | 28 | Capitan Bado | 19-Apr-2022 | 15-Apr-2022 | M | 52 | 71 |
| OQ567808 | DENV-1 | Genotype V | PY | 26 | Capitan Bado | 18-Apr-2022 | 16-Apr-2022 | M | 20 | 75.1 |
| OQ567809 | DENV-1 | Genotype V | PY | 21 | Capitan Bado | 21-Apr-2022 | 19-Apr-2022 | F | 11 | 83.9 |
| OQ567810 | DENV-1 | Genotype V | PY | 28 | Capitan Bado | 22-Apr-2022 | 20-Apr-2022 | M | 36 | 88.8 |
| OQ567811 | DENV-1 | Genotype V | PY | 22 | Capitan Bado | 21-Apr-2022 | 17-Apr-2022 | M | 61 | 93.8 |
| OQ567812 | DENV-1 | Genotype V | PY | 26 | Capitan Bado | 20-Apr-2022 | 16-Apr-2022 | M | 33 | 94.2 |
| OQ567813 | DENV-1 | Genotype V | PY | 27 | Capitan Bado | 20-Apr-2022 | 17-Apr-2022 | M | 61 | 93.4 |
| OQ567814 | DENV-1 | Genotype V | PY | 21 | Encarnacion | 4-May-2022 | 2-May-2022 | F | 18 | 94.2 |
| OQ567815 | DENV-1 | Genotype V | PY | 21 | Villa Ygatimi | 10-May-2022 | 7-May-2022 | F | 62 | 88.3 |
| OQ567816 | DENV-1 | Genotype V | PY | 24 | Capitan Bado | 4-May-2022 | 2-May-2022 | F | 18 | 79.4 |
| OQ567817 | DENV-1 | Genotype V | PY | 27 | Pedro Juan Caballero | 5-Jun-2022 | 1-Jun-2022 | F | 37 | 93.5 |
| OQ567818 | DENV-1 | Genotype V | PY | 27 | Salto del Guaira | 2-Jun-2022 | 1-Jun-2022 | F | 14 | 94.2 |
| OQ567819 | DENV-1 | Genotype V | PY | 22 | Salto del Guaira | 20-Jun-2022 | 16-Jun-2022 | M | 72 | 93 |
| OQ567820 | DENV-1 | Genotype V | PY | 25 | Capitan Bado | 14-Jun-2022 | 11-Jun-2022 | F | 17 | 84.7 |
| OQ567821 | DENV-2 | Genotype III | PY | 26 | Villarrica | 2-Jul-2021 | 1-Jul-2021 | M | 29 | 75.3 |
| OQ567822 | DENV-2 | Genotype III | PY | 26 | Mariano Roque Alonso | 10-Jul-2021 | 10-Jul-2021 | F | 21 | 70 |
| OQ567823 | DENV-2 | Genotype III | PY | 28 | Coronel Oviedo | 28-Mar-2022 | 26-Mar-2022 | M | 35 | 90.9 |
| OQ567824 | DENV-2 | Genotype III | PY | 26 | Coronel Oviedo | 27-Mar-2022 | 24-Mar-2022 | M | 49 | 93.1 |
| OQ567825 | DENV-2 | Genotype III | PY | 21 | Coronel Oviedo | 29-Mar-2022 | 27-Mar-2022 | M | 23 | 91.1 |
| OQ567826 | DENV-2 | Genotype III | PY | 28 | Coronel Oviedo | 5-Apr-2022 | 2-Apr-2022 | M | 28 | 93.1 |
| OQ567827 | DENV-2 | Genotype III | PY | 22 | Coronel Oviedo | 11-Apr-2022 | 9-Apr-2022 | F | 33 | 87.2 |
| OQ567828 | DENV-2 | Genotype III | PY | 26 | Coronel Oviedo | 7-Apr-2022 | 5-Apr-2022 | F | 29 | 78.6 |
| OQ567829 | DENV-2 | Genotype III | PY | 27 | Coronel Oviedo | 17-Apr-2022 | 14-Apr-2022 | M | 22 | 93 |
| OQ567830 | DENV-2 | Genotype III | PY | 21 | Coronel Oviedo | 18-Apr-2022 | 16-Apr-2022 | F | 48 | 93.2 |
| OQ567831 | DENV-2 | Genotype III | PY | 21 | Caaguazu | 22-Apr-2022 | 20-Apr-2022 | M | 61 | 93.6 |
| OQ567832 | DENV-2 | Genotype III | PY | 24 | Coronel Oviedo | 28-Apr-2022 | 26-Apr-2022 | F | 45 | 71.1 |
| OQ567833 | DENV-2 | Genotype III | PY | 27 | Coronel Oviedo | 2-May-2022 | 1-May-2022 | M | 51 | 93.3 |
| OQ567834 | DENV-2 | Genotype III | PY | 27 | Coronel Oviedo | 27-Apr-2022 | 25-Apr-2022 | M | 59 | 84.4 |
| OQ567835 | DENV-2 | Genotype III | PY | 22 | Coronel Oviedo | 6-May-2022 | 3-May-2022 | F | 42 | 90.7 |
| OQ567836 | DENV-2 | Genotype III | PY | 25 | Coronel Oviedo | 9-May-2022 | 5-May-2022 | M | 51 | 89.8 |
| OQ567837 | DENV-2 | Genotype III | PY | 26 | Coronel Oviedo | 8-May-2022 | 6-May-2022 | F | 43 | 86.8 |
| OQ567838 | DENV-2 | Genotype III | PY | 26 | Coronel Oviedo | 22-May-2022 | 18-May-2022 | F | 37 | 89.8 |
| OQ567839 | DENV-2 | Genotype III | PY | 28 | Coronel Oviedo | 29-May-2022 | 19-May-2022 | F | 26 | 92 |
| OQ567840 | DENV-2 | Genotype III | PY | 26 | Repatriacion | 24-Jun-2022 | 23-Jun-2022 | F | 44 | 88.4 |
| OQ567841 | DENV-2 | Genotype III | PY | 21 | Benjamin Aceval | 23-Feb-2022 | 22-Feb-2022 | M | 14 | 78.8 |
| OQ567842 | DENV-2 | Genotype III | PY | 28 | Coronel Oviedo | 22-Mar-2022 | 20-Mar-2022 | F | 46 | 92.9 |
| OQ567843 | DENV-2 | Genotype III | PY | 22 | Coronel Oviedo | 5-May-2022 | 4-May-2022 | M | 12 | 70.2 |
| OQ567844 | DENV-2 | Genotype III | PY | 26 | Coronel Oviedo | 4-May-2022 | 4-May-2022 | M | 24 | 93.2 |
| OQ567845 | DENV-2 | Genotype III | PY | 27 | Capiata | 27-Apr-2022 | 24-Apr-2022 | F | 20 | 87.2 |
| OQ567846 | DENV-2 | Genotype III | PY | 21 | Presidente Franco | 24-Mar-2021 | 22-Mar-2021 | F | 61 | 92.8 |
| OQ567847 | DENV-2 | Genotype III | PY | 21 | Encarnacion | 14-Apr-2021 | 4-Dec-2021 | M | 59 | 92.7 |
| OQ567848 | DENV-4 | Genotype II | PY | 24 | Coronel Oviedo | 7-Mar-2016 | 7-Mar-2016 | F | 37 | 91.9 |
| OQ567849 | DENV-4 | Genotype II | PY | 27 | Asuncion | 27-Jan-2020 | 20-Jan-2024 | F | 47 | 92.2 |
| OQ567850 | DENV-4 | Genotype II | PY | 24 | Asuncion | 30-Jan-2020 | 28-Jan-2020 | F | 14 | 92 |
| OQ567851 | DENV-4 | Genotype II | PY | 28 | Filadelfia | 1-Feb-2020 | 1-Feb-2020 | M | 11 | 92 |
| OQ567852 | DENV-4 | Genotype II | PY | 20 | Lambare | 4-Feb-2020 | 2-Apr-2020 | M | 29 | 92.5 |
| OQ567853 | DENV-4 | Genotype II | PY | 27 | Asuncion | 6-Feb-2020 | 4-Feb-2020 | M | 30 | 91.7 |
| OQ567854 | DENV-4 | Genotype II | PY | 23 | Pilar | 4-Feb-2020 | 4-Feb-2020 | F | 60 | 92 |
| OQ567855 | DENV-4 | Genotype II | PY | 26 | Itaugua | 7-Feb-2020 | 3-Feb-2020 | F | 40 | 92.5 |
| OQ567856 | DENV-4 | Genotype II | PY | 26 | Itagua | 10-Feb-2020 | 8-Feb-2020 | F | 77 | 92.5 |
| OQ567857 | DENV-4 | Genotype II | PY | 29 | Obligado | 13-Feb-2020 | 2-Nov-2020 | F | 4 | 92 |
| OQ567858 | DENV-4 | Genotype II | PY | 26 | Itagua | 1-Jan-2020 | 31-Dec-2019 | F | 29 | 92 |
| OQ567859 | DENV-4 | Genotype II | PY | 21 | Lambare | 7-Jan-2020 | 1-May-2020 | M | 31 | 93 |
| OQ567860 | DENV-4 | Genotype II | PY | 25 | Villa Elisa | 7-Jan-2020 | 1-Apr-2020 | F | 32 | 92.6 |
| OQ567861 | DENV-4 | Genotype II | PY | 17 | Nemby | 9-Jan-2020 | 1-Sep-2020 | M | 1M | 92.5 |
| OQ567862 | DENV-4 | Genotype II | PY | 27 | Itagua | 12-Jan-2020 | 1-Nov-2020 | F | 30 | 91.8 |
| OQ567863 | DENV-4 | Genotype II | PY | 30 | Nemby | 13-Jan-2020 | 1-Dec-2020 | F | 29 | 91.9 |
| OQ567864 | DENV-4 | Genotype II | PY | 21 | Trinidad | 13-Jan-2020 | 13-Jan-2020 | M | 47 | 92.4 |
| OQ567865 | DENV-4 | Genotype II | PY | 25 | Coronel Oviedo | 11-Jan-2020 | 1-Nov-2020 | M | 59 | 92.6 |
| OQ567866 | DENV-4 | Genotype II | PY | 30 | San Lorenzo | 12-Jan-2020 | 1-Dec-2020 | F | 77 | 92 |
| OQ567867 | DENV-4 | Genotype II | PY | 23 | San Lorenzo | 17-Jan-2020 | 15-Jan-2020 | F | 37 | 92.4 |
| OQ567868 | DENV-4 | Genotype II | PY | 23 | Ypane | 18-Jan-2020 | 12-Jan-2020 | M | 25 | 92.1 |
| OQ567869 | DENV-4 | Genotype II | PY | 19 | San Lorenzo | 15-Jan-2020 | 11-Jan-2020 | M | 5M | 92.7 |
| OQ567870 | DENV-4 | Genotype II | PY | 26 | Itagua | 17-Jan-2020 | 12-Jan-2020 | F | 18 | 92.9 |
| OQ567871 | DENV-4 | Genotype II | PY | 20 | San Lorenzo | 20-Jan-2020 | 15-Jan-2020 | F | 1 | 92.5 |
| OQ567872 | DENV-4 | Genotype II | PY | 30 | Pilar | 22-Feb-2021 | 19-Feb-2021 | F | 7 | 92.1 |
| Variable 1 | Variable 2 | Correlation (Spearman’s Coefficient) |
|---|---|---|
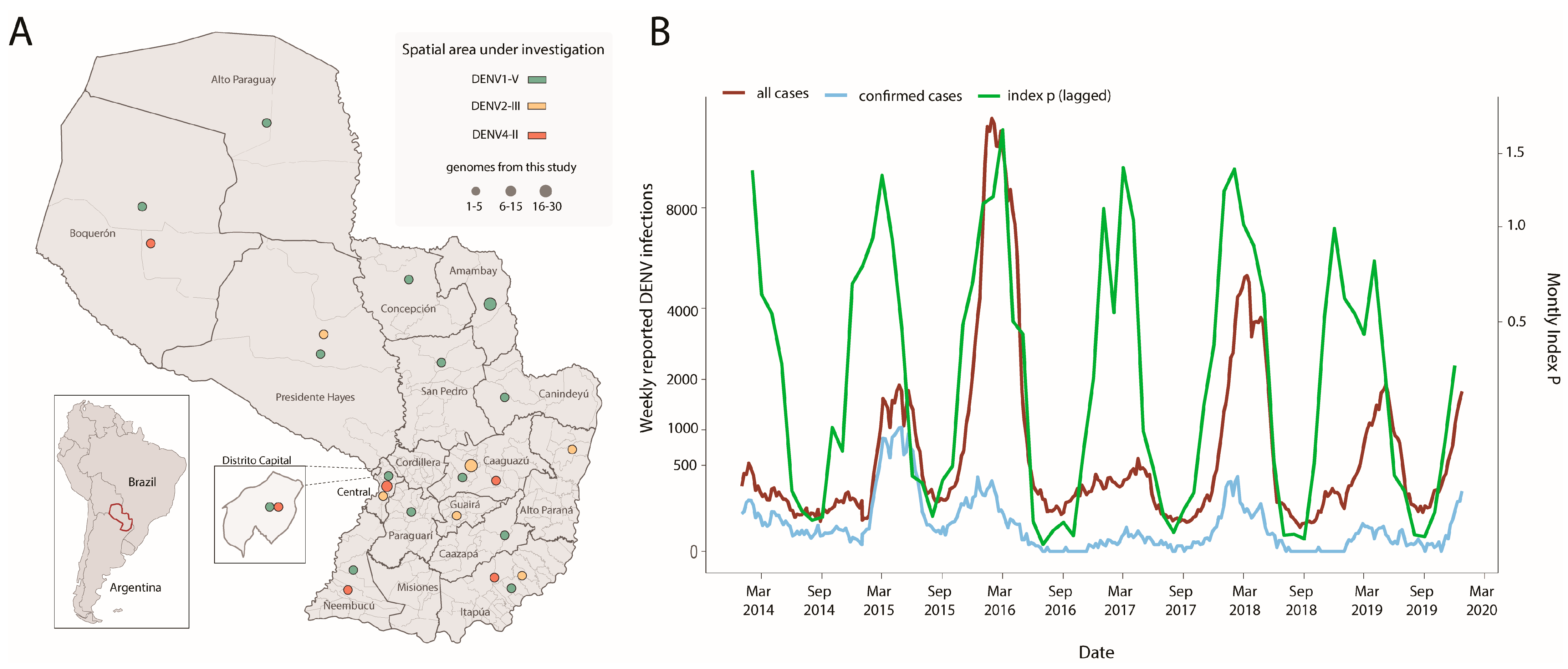
| index p | total cases * | 0.55 |
| temperature | total cases * | 0.37 |
| humidity | total cases * | 0.50 |
| precipitation | total cases * | 0.40 |
| index p | confirmed cases | 0.51 |
| temperature | confirmed cases | 0.34 |
| humidity | confirmed cases | 0.43 |
| precipitation | confirmed cases | 0.42 |
| index p (lagged) | total cases * | 0.71 |
| index p (lagged) | confirmed cases | 0.62 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vazquez, C.; Alcantara, L.C.J.; Fonseca, V.; Lima, M.; Xavier, J.; Adelino, T.; Fritsch, H.; Castro, E.; de Oliveira, C.; Schuab, G.; et al. Retrospective Spatio-Temporal Dynamics of Dengue Virus 1, 2 and 4 in Paraguay. Viruses 2023, 15, 1275. https://doi.org/10.3390/v15061275
Vazquez C, Alcantara LCJ, Fonseca V, Lima M, Xavier J, Adelino T, Fritsch H, Castro E, de Oliveira C, Schuab G, et al. Retrospective Spatio-Temporal Dynamics of Dengue Virus 1, 2 and 4 in Paraguay. Viruses. 2023; 15(6):1275. https://doi.org/10.3390/v15061275
Chicago/Turabian StyleVazquez, Cynthia, Luiz Carlos Junior Alcantara, Vagner Fonseca, Mauricio Lima, Joilson Xavier, Talita Adelino, Hegger Fritsch, Emerson Castro, Carla de Oliveira, Gabriel Schuab, and et al. 2023. "Retrospective Spatio-Temporal Dynamics of Dengue Virus 1, 2 and 4 in Paraguay" Viruses 15, no. 6: 1275. https://doi.org/10.3390/v15061275
APA StyleVazquez, C., Alcantara, L. C. J., Fonseca, V., Lima, M., Xavier, J., Adelino, T., Fritsch, H., Castro, E., de Oliveira, C., Schuab, G., Lima, A. R. J., Villalba, S., Gomez de la Fuente, A., Rojas, A., Cantero, C., Fleitas, F., Aquino, C., Ojeda, A., Sequera, G., ... Giovanetti, M. (2023). Retrospective Spatio-Temporal Dynamics of Dengue Virus 1, 2 and 4 in Paraguay. Viruses, 15(6), 1275. https://doi.org/10.3390/v15061275