

Genetic Diversity and Dispersal of DENGUE Virus among Three Main Island Groups of the Philippines during 2015–2017
 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study
2.2. Sample Collection
2.3. Serotyping of Dengue Virus
2.4. Reverse Transcription-Polymerase Chain Reaction Amplification and Sequencing of Envelope Gene of Dengue Virus
2.5. Assembly and Comparison of Nucleotide Sequences
2.6. Selection Pressure Analysis
2.7. Phylogenetic Analysis of Envelope Gene Sequences
2.8. Phylogeography Analysis of Envelope Gene Sequences
3. Results
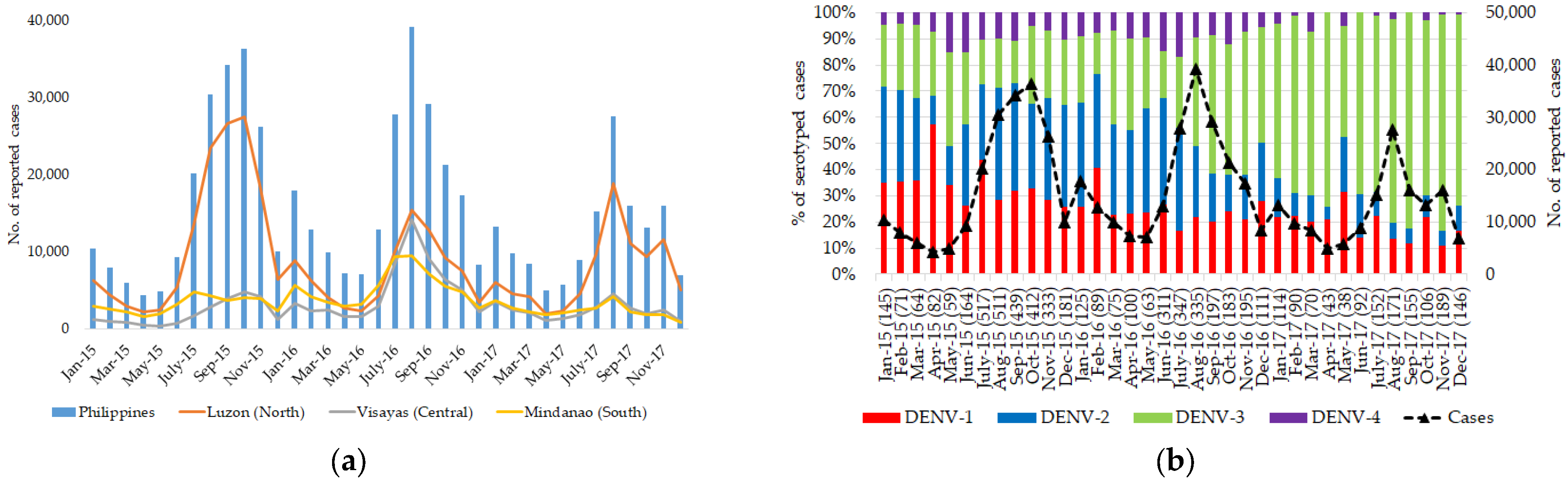
3.1. Dengue Is Widespread and Hyperendemic in the Philippines
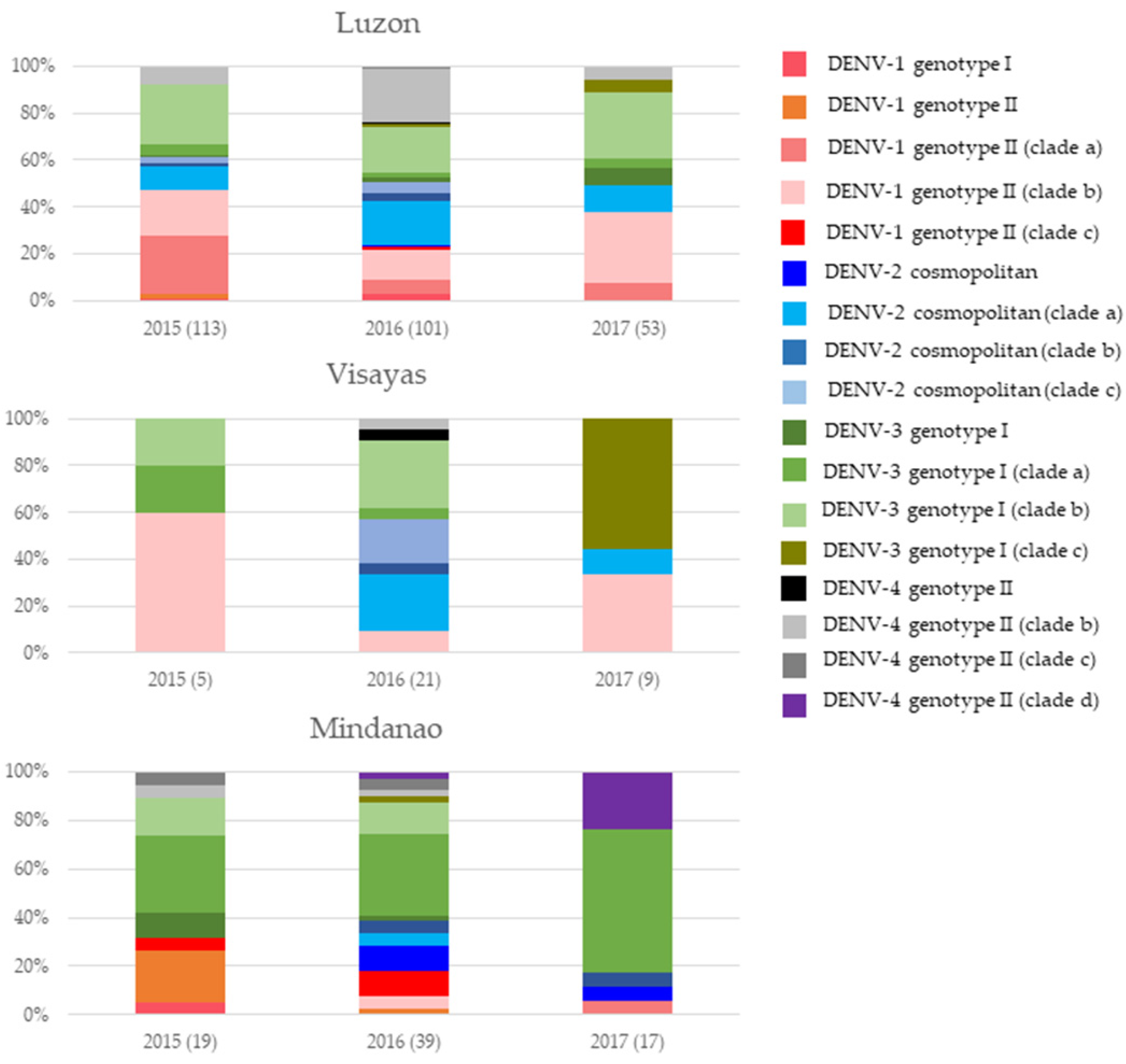
3.2. DENV Serotypes Consisted of Multiple Clades That Constituted Distinct Genotype Compositions in Each Island Group
3.3. Luzon Island Is the Most Probable Hub of DENV Dispersal in the Philippines
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guo, C.; Zhou, Z.; Wen, Z.; Liu, Y.; Zeng, C.; Xiao, D.; Ou, M.; Han, Y.; Huang, S.; Liu, D.; et al. Global Epidemiology of Dengue Outbreaks in 1990–2015: A Systematic Review and Meta-Analysis. Front. Cell. Infect. Microbiol. 2017, 7, 317. [Google Scholar] [CrossRef] [PubMed]
- Togami, E.; Chiew, M.; Lowbridge, C.; Biaukula, V.; Bell, L.; Yajima, A.; Eshofonie, A.; Saulo, D.; Hien, D.T.H.; Otsu, S.; et al. Epidemiology of dengue reported in the World Health Organization’s Western Pacific Region, 2013–2019. West. Pac. Surveill. Response J. WPSAR 2023, 14, 1–16. [Google Scholar]
- Siler, J.F.; Hall, M.W.; Hitchens, A.P. Dengue: Its history, epidemiology, mechanism of transmission, etiology, clinical manifestations, immunity, and prevention. Philipp. J. Sci. 1926, 29, 1–304. [Google Scholar]
- Ooi, E.-E.; Gubler, D.J. Dengue in Southeast Asia: Epidemiological characteristics and strategic challenges in disease prevention. Cad. De Saude Publica 2009, 25, S115–S124. [Google Scholar] [CrossRef]
- Gubler, D. Dengue and Dengue Haemorrhagic Fever its History and Resurgence as Aglobal Public Health Problem Dalam DJ Gubler and G. Dengue and Dengue Haemorrhagic Fever; CAB International: London, UK, 1997. [Google Scholar]
- Hayes, C.G.; Manaloto, C.R.; Gonzales, A.; Ranoa, C.P. Dengue Infections in the Philippines: Clinical and Virological Findings in 517 Hospitalized Patients. Am. J. Trop. Med. Hyg. 1988, 39, 110–116. [Google Scholar] [CrossRef]
- Songco, R.S.; Hayes, C.G.; Leus, C.D.; Manaloto, C.O. Dengue fever/dengue haemorrhagic fever in Filipino children: Clinical experience during the 1983-1984 epidemic. Southeast Asian J. Trop. Med. Public Health 1987, 18, 284–290. [Google Scholar]
- Venzon, E.; Rudnick, A.; Marchette, N.; Fabie, A.; Dukellis, E. The Greater Manila dengue hemorrhagic fever epidemic of 1966. J. Philipp. Isl. Med. Assoc. 1972, 48, 297–313. [Google Scholar]
- Matias, R. A historical review of dengue virus research in the Philippines. St. Luke’s J. Med. 2003, 1, 3–8. [Google Scholar]
- Mahilum, M.M.; Ludwig, M.; Madon, M.B.; Becker, N. Evaluation of the present dengue situation and control strategies against Aedes aegypti in Cebu City, Philippines. J. Vector Ecol. 2005, 30, 277–283. [Google Scholar]
- Lewis, J.A.; Chang, G.-J.; Lanciotti, R.S.; Kinney, R.M.; Mayer, L.W.; Trent, D.W. Phylogenetic Relationships of Dengue-2 Viruses. Virology 1993, 197, 216–224. [Google Scholar] [CrossRef]
- Lanciotti, R.S.; Lewis, J.G.; Gubler, D.J.; Trent, D.W. Molecular evolution and epidemiology of dengue-3 viruses. J. Gen. Virol. 1994, 75, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Lanciotti, R.S.; Trent, D.W.; Gubler, D.J. Molecular evolution and phylogeny of dengue-4 viruses. J. Gen. Virol. 1997, 78, 2279–2284. [Google Scholar] [CrossRef] [PubMed]
- Rico-Hesse, R.; Harrison, L.M.; Salas, R.A.; Tovar, D.; Nisalak, A.; Ramos, C.; Boshell, J.; de Mesa, M.T.R.; Nogueira, R.M.; da Rosa, A.T. Origins of dengue type 2 viruses associated with increased pathogenicity in the Americas. Virology 1997, 230, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Rico-Hesse, R.; Harrison, L.M.; Nisalak, A.; Vaughn, D.W.; Kalayanarooj, S.; Green, S.; Rothman, A.L.; Ennis, F.A. Molecular evolution of dengue type 2 virus in Thailand. Am. J. Trop. Med. Hyg. 1998, 58, 96–101. [Google Scholar] [CrossRef]
- Gubler, D.; Suharyono, W.; Lubis, I.; Eram, S.; Gunarso, S. Epidemic dengue 3 in central Java, associated with low viremia in man. Am. J. Trop. Med. Hyg. 1981, 30, 1094–1099. [Google Scholar] [CrossRef]
- Leitmeyer, K.C.; Vaughn, D.W.; Watts, D.M.; Salas, R.; Villalobos, I.; Chacon, D.; Ramos, C.; Rico-Hesse, R. Dengue Virus Structural Differences That Correlate with Pathogenesis. J. Virol. 1999, 73, 4738–4747. [Google Scholar] [CrossRef]
- Salda, L.T.D.; Natividad, F.F.; Morita, K.; Matias, R.R.; Parquet, M.D.C.; Kobayashi, N. Molecular epidemiology of dengue 2 viruses in the philippines: Genotype shift and local evolution. Am. J. Trop. Med. Hyg. 2005, 73, 796–802. [Google Scholar] [CrossRef]
- Usme-Ciro, J.A.; Méndez, J.A.; Laiton, K.D.; Páez, A. The relevance of dengue virus genotypes surveillance at country level before vaccine approval. Hum. Vaccines Immunother. 2014, 10, 2674–2678. [Google Scholar] [CrossRef]
- Lee, K.-S.; Lai, Y.-L.; Lo, S.; Barkham, T.; Aw, P.; Ooi, P.-L.; Tai, J.-C.; Hibberd, M.; Johansson, P.; Khoo, S.-P.; et al. Dengue Virus Surveillance for Early Warning, Singapore. Emerg. Infect. Dis. 2010, 16, 847–849. [Google Scholar] [CrossRef]
- Agrupis, K.A.; Ylade, M.; Aldaba, J.; Lopez, A.L.; Deen, J. Trends in dengue research in the Philippines: A systematic review. PLoS Neglected Trop. Dis. 2019, 13, e0007280. [Google Scholar] [CrossRef]
- Galarion, M.J.; Schwem, B.; Pangilinan, C.; Tonga, A.D.; Petronio-Santos, J.A.; Reyes, E.D.; Destura, R. Genotypic persistence of dengue virus in the Philippines. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2019, 69, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Li, L.S. National dengue prevention and control program in the philippines. In Proceedings of the Asia-Pacific Dengue Program Managers Meeting, Singapore, 5–8 May 2008; World Health Organization—Western Pacific Region: Manila, Philippines, 2008; pp. 94–98. [Google Scholar]
- Bravo, L.; Roque, V.G.; Brett, J.; Dizon, R.; L’Azou, M. Epidemiology of Dengue Disease in the Philippines (2000–2011): A Systematic Literature Review. PLoS Negl. Trop. Dis. 2014, 8, e3027. [Google Scholar] [CrossRef] [PubMed]
- WHO. Dengue: Guidelines for Diagnosis, Treatment, Prevention and Control; WHO: Geneva, Switzerland, 2009. [Google Scholar]
- Johnson, B.W.; Russell, B.J.; Lanciotti, R.S. Serotype-Specific Detection of Dengue Viruses in a Fourplex Real-Time Reverse Transcriptase PCR Assay. J. Clin. Microbiol. 2005, 43, 4977–4983. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Delport, W.; Poon, A.F.Y.; Frost, S.D.W.; Pond, S.L.K. Datamonkey 2010: A suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 2010, 26, 2455–2457. [Google Scholar] [CrossRef] [PubMed]
- Koo, C.; Tien, W.P.; Xu, H.; Ong, J.; Rajarethinam, J.; Lai, Y.L.; Ng, L.-C.; Hapuarachchi, H.C. Highly Selective Transmission Success of Dengue Virus Type 1 Lineages in a Dynamic Virus Population: An Evolutionary and Fitness Perspective. iScience 2018, 6, 38–51. [Google Scholar] [CrossRef]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Drummond, A.J.; Ho, S.Y.W.; Phillips, M.J.; Rambaut, A. Relaxed Phylogenetics and Dating with Confidence. PLoS Biol. 2006, 4, e88. [Google Scholar] [CrossRef]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian Phylogeography Finds Its Roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A. Tracer: A Program for Analysing Results from Bayesian MCMC Programs such as BEAST & MrBayes; University of Edinburgh: Edinburgh, UK, 2003. [Google Scholar]
- Bielejec, F.; Rambaut, A.; Suchard, M.A.; Lemey, P. SPREAD: Spatial phylogenetic reconstruction of evolutionary dynamics. Bioinformatics 2011, 27, 2910–2912. [Google Scholar] [CrossRef] [PubMed]
- Faria, N.R.; Suchard, M.A.; Rambaut, A.; Streicker, D.G.; Lemey, P. Simultaneously reconstructing viral cross-species transmission history and identifying the underlying constraints. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20120196. [Google Scholar] [CrossRef] [PubMed]
- Su, G.L.S. Correlation of Climatic Factors and Dengue Incidence in Metro Manila, Philippines. AMBIO A J. Hum. Environ. 2008, 37, 292–294. [Google Scholar] [CrossRef]
- Epidemiology Bureau. Epidemic-Prone Disease Case Surveillance. Annual Report. 2021; pp. 74–79. Available online: https://doh.gov.ph/statistics (accessed on 1 March 2023).
- Ong, E.P.; Obeles, A.J.T.; Ong, B.A.G.; Tantengco, O.A.G. Perspectives and lessons from the Philippines’ decades-long battle with dengue. Lancet Reg. Health West. Pac. 2022, 24, 100505. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, N. Current DF/DHF Prevention and Control Programme in the Philippines. Dengue Bull. 1997, 21, 41–46. [Google Scholar]
- Rajarethinam, J.; Ang, L.W.; Ong, J.; Ycasas, J.; Hapuarachchi, H.C.; Yap, G.; Chong, C.-S.; Lai, Y.-L.; Cutter, J.; Ho, D.; et al. Dengue in Singapore from 2004 to 2016: Cyclical Epidemic Patterns Dominated by Serotypes 1 and 2. Am. J. Trop. Med. Hyg. 2018, 99, 204–210. [Google Scholar] [CrossRef]
- Jagtap, S.; Pattabiraman, C.; Sankaradoss, A.; Krishna, S.; Roy, R. Evolutionary dynamics of dengue virus in India. PLoS Pathog. 2023, 19, e1010862. [Google Scholar] [CrossRef]
- Lee, K.-S.; Lo, S.; Tan, S.S.-Y.; Chua, R.; Tan, L.-K.; Xu, H.; Ng, L.-C. Dengue virus surveillance in Singapore reveals high viral diversity through multiple introductions and in situ evolution. Infect. Genet. Evol. 2012, 12, 77–85. [Google Scholar] [CrossRef]
- Petronio, J.A.G.; Vinarao, R.B.; Flores, K.M.G.; Destura, R.V. Continued circulation of a single genotype of dengue virus serotype 2 in the Philippines. Asian Pac. J. Trop. Med. 2014, 7, 30–33. [Google Scholar] [CrossRef]
- Luz, M.A.D.V.; Nabeshima, T.; Moi, M.L.; Dimamay, M.T.A.; Pangilinan, L.-A.S.; Dimamay, M.P.S.; Matias, R.R.; Mapua, C.A.; Buerano, C.C.; De Guzman, F.; et al. An Epidemic of Dengue Virus Serotype-4 during the 2015–2017: The Emergence of a Novel Genotype IIa of DENV-4 in the Philippines. Jpn. J. Infect. Dis. 2020, 73, 176. [Google Scholar] [CrossRef]
- Harapan, H.; Michie, A.; Sasmono, R.T.; Imrie, A. Dengue: A Minireview. Viruses 2020, 12, 829. [Google Scholar] [CrossRef] [PubMed]
- Dolan, P.T.; Taguwa, S.; Rangel, M.A.; Acevedo, A.; Hagai, T.; Andino, R.; Frydman, J. Principles of dengue virus evolvability derived from genotype-fitness maps in human and mosquito cells. eLife 2021, 10, e61921. [Google Scholar] [CrossRef] [PubMed]
- Holmes, E.C. Patterns of Intra- and Interhost Nonsynonymous Variation Reveal Strong Purifying Selection in Dengue Virus. J. Virol. 2003, 77, 11296–11298. [Google Scholar] [CrossRef] [PubMed]
- Woelk, C.H.; Holmes, E.C. Reduced Positive Selection in Vector-Borne RNA Viruses. Mol. Biol. Evol. 2002, 19, 2333–2336. [Google Scholar] [CrossRef] [PubMed]
- Lequime, S.; Fontaine, A.; Gouilh, M.A.; Moltini-Conclois, I.; Lambrechts, L. Genetic Drift, Purifying Selection and Vector Genotype Shape Dengue Virus Intra-host Genetic Diversity in Mosquitoes. PLoS Genet. 2016, 12, e1006111. [Google Scholar] [CrossRef] [PubMed]
- Reiter, P.; Clark, G.G.; Anderson, R.A.; Amador, M.A. Short Report: Dispersal of Aedes aegypti in an Urban Area after Blood Feeding as Demonstrated by Rubidium-Marked Eggs. Am. J. Trop. Med. Hyg. 1995, 52, 177–179. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Serotype | DENV Strains | Genetic Similarity (Nucleotide and Amino Acid Level) | Mean Evolutionary Rate (95% HPD) | tMRCA (95% HPD) | Transmission Period |
|---|---|---|---|---|---|
| DENV-1 | Genotype IIa | 99.1–100% (98.9–100%) | 8.6 (3.7–14.3) | 4.8 (3.6–6.6) | June 2015–May 2017 |
| Genotype IIb | 97.7–100% (98.9–100%) | 10.3 (4.8–17.0) | 13.3 (10.7–16.4) | January 2015–May 2017 | |
| Genotype IIc | 99.5–99.9% (99.9–100%) | 5.4 (2.2–9.1) | 4.0 (2.7–7.3) | November 2015–November 2016 | |
| DENV-2 | Cosmopolitan genotype a | 98–99.9% (98.5–100%) | 12.5 (4.2–22.9) | 8.4 (6.4–10.7) | March 2015–March 2017 |
| Cosmopolitan genotype b | 99.0–100% (99.1–100%) | 10.3 (4.6–17.6) | 5.2 (3.5–7.2) | July 2015–April 2017 | |
| Cosmopolitan genotype c | 98.6–100% (98.9–100%) | 8.5 (2.2–16.7) | 7.1 (5.0–9.7) | August 2015–August 2016 | |
| DENV-3 | Genotype Ia | 98.4–100% (98.9–100%) | 8.5 (2.1–17.8) | 8.4 (4.4–9.0) | June 2015–April 2017 |
| Genotype Ib | 98–100% (99.1–100%) | 7.9 (1.8–16.6) | 6.4 (5.7–11.7) | March 2015–May 2017 | |
| Genotype Ic | 98.7–99.9% (99.5–100%) | 6.2 (1.4–12.5) | 5.4 (3.1–8.3) | September 2016–April 2017 | |
| DENV-4 | Genotype IIb | 98.5–100% (99.1–100%) | 8.0 (1.1–17.7) | 7.0 (4.9–9.7) | August 2015–February 2017 |
| Genotype IIc | 99.5–99.9% (99.1–100%) | 11.1 (2.4–23.7) | 3.7 (2.2–5.9) | November 2015–December 2016 | |
| Genotype IId | 99.2–99.7% (99.3–100%) | 4.5 (1.6–8.0) | 3 (1.2–5.7) | September 2016–May 2017 |
| Island Group | Region | No. of Dispersal Pathways £ | |||
|---|---|---|---|---|---|
| DENV-1 | DENV-2 | DENV-3 | DENV-4 | ||
| Luzon | llocos Region (I) | 0 | 1 | 0 | 0 |
| Cagayan Valley (II) | 2 | 0 | 0 | 0 | |
| Central Luzon (III) | 1 | 4 | 1 | 0 | |
| Calabarzon (IVA) | 4 | 3 | 1 | 1 | |
| Mimaropa Region (XVII) | 2 | 2 | 1 | 1 | |
| Bicol Region (V) | 0 | 0 | 1 | 0 | |
| CAR (XV) | 3 | 4 | 1 | 0 | |
| NCR (XIV) | 1 | 2 | 3 | 0 | |
| Visayas | Western Visayas (VI) | 0 | 0 | 3 | 1 |
| Central Visayas (VII) | 0 | 0 | 1 | 0 | |
| Eastern Visayas (VIII) | 1 | 1 | 0 | 0 | |
| Mindanao | Zamboanga Peninsula (IX) | 0 | 0 | 0 | 1 |
| Northern Mindanao (X) | 0 | 0 | 0 | 0 | |
| Davao Region (XI) | 3 | 0 | 2 | 0 | |
| SOCCSKSARGEN (XII) | 1 | 0 | 2 | 1 | |
| Caraga Region (XIII) | 2 | 0 | 3 | 3 | |
| ARMM (XVI) | 0 | 0 | 1 | 0 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sy, A.K.; Koo, C.; Privaldos, K.J.R.; Quinones, M.A.T.; Igoy, M.A.U.; Villanueva, S.Y.A.M.; Hibberd, M.L.; Ng, L.C.; Hapuarachchi, H.C. Genetic Diversity and Dispersal of DENGUE Virus among Three Main Island Groups of the Philippines during 2015–2017. Viruses 2023, 15, 1079. https://doi.org/10.3390/v15051079
Sy AK, Koo C, Privaldos KJR, Quinones MAT, Igoy MAU, Villanueva SYAM, Hibberd ML, Ng LC, Hapuarachchi HC. Genetic Diversity and Dispersal of DENGUE Virus among Three Main Island Groups of the Philippines during 2015–2017. Viruses. 2023; 15(5):1079. https://doi.org/10.3390/v15051079
Chicago/Turabian StyleSy, Ava Kristy, Carmen Koo, Kristine J. R. Privaldos, Mary Ann T. Quinones, Mary A. U. Igoy, Sharon Y. A. M. Villanueva, Martin L. Hibberd, Lee Ching Ng, and Hapuarachchige C. Hapuarachchi. 2023. "Genetic Diversity and Dispersal of DENGUE Virus among Three Main Island Groups of the Philippines during 2015–2017" Viruses 15, no. 5: 1079. https://doi.org/10.3390/v15051079
APA StyleSy, A. K., Koo, C., Privaldos, K. J. R., Quinones, M. A. T., Igoy, M. A. U., Villanueva, S. Y. A. M., Hibberd, M. L., Ng, L. C., & Hapuarachchi, H. C. (2023). Genetic Diversity and Dispersal of DENGUE Virus among Three Main Island Groups of the Philippines during 2015–2017. Viruses, 15(5), 1079. https://doi.org/10.3390/v15051079