
An Artificial Peptide-Based Bifunctional HIV-1 Entry Inhibitor That Interferes with Viral Glycoprotein-41 Six-Helix Bundle Formation and Antagonizes CCR5 on the Host Cell Membrane

 ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
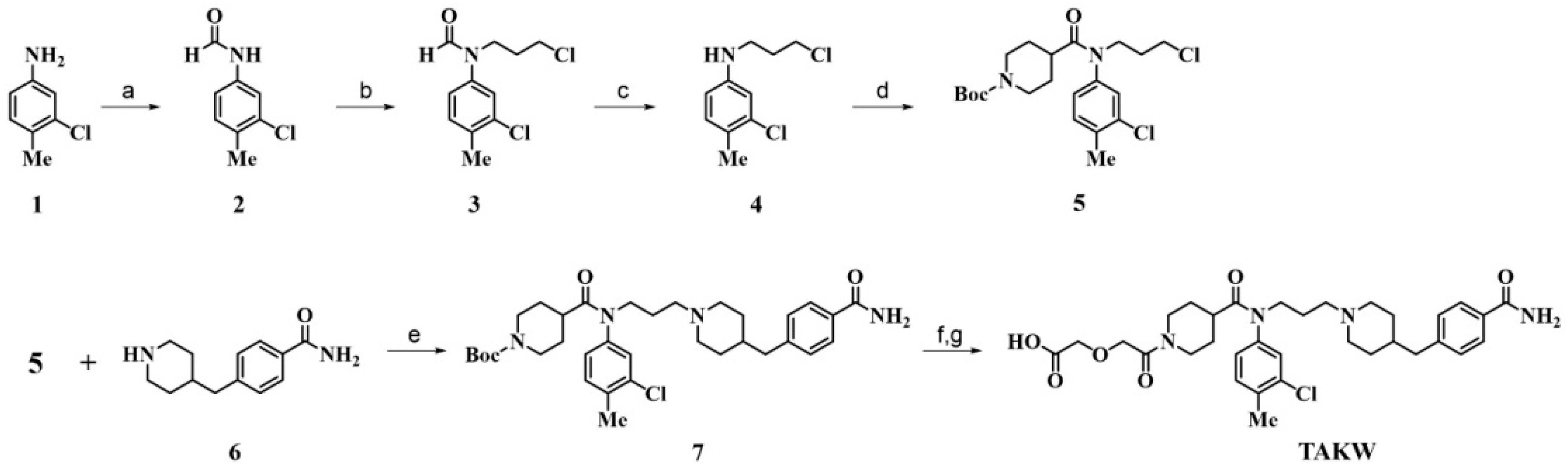
2.1. Chemistry
2.2. Peptide Synthesis
2.3. HIV-1 Infection Assay
2.4. Cytotoxicity Assay
2.5. Circular Dichroism (CD) Spectroscopy
2.6. Native Polyacrylamide Gel Electrophoresis (N-PAGE)
2.7. Calcium Mobilization Assay
2.8. Solubility
2.9. Metabolic Stability
3. Results and Discussion
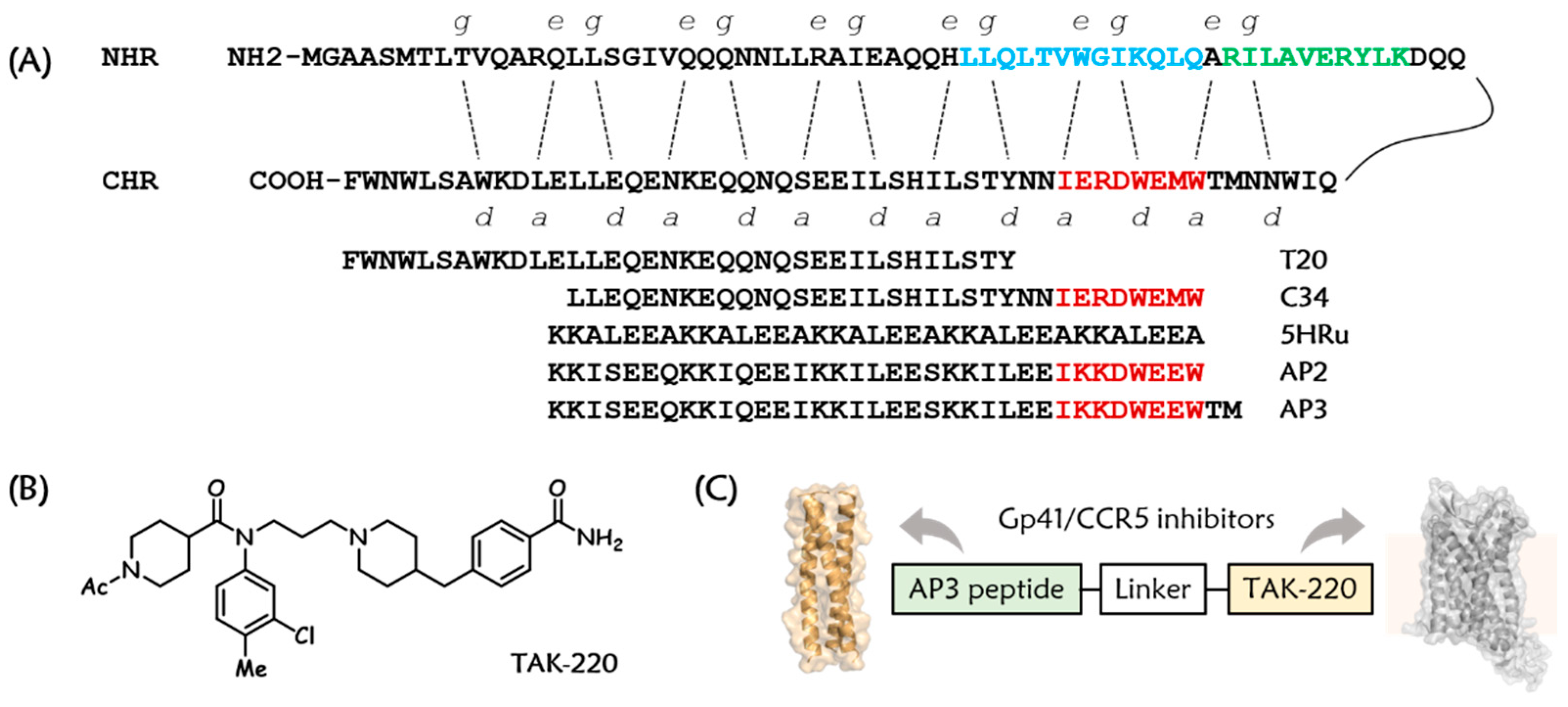
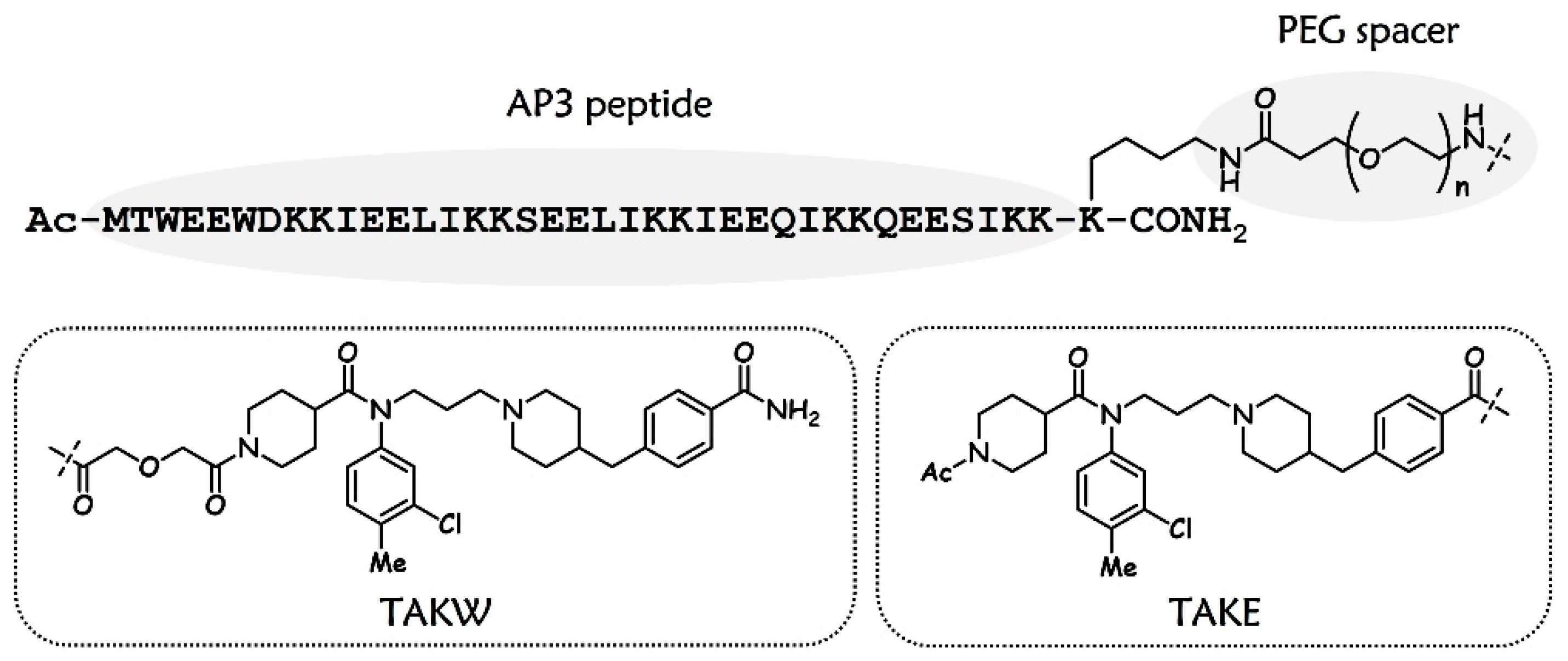
3.1. Design and Chemistry
3.2. Potent Antiviral Activity of Artificially Designed Peptides against HIV-1 Strains
3.3. Antiviral Activities of AP3P4E against T20-Resistant HIV-1 Variants
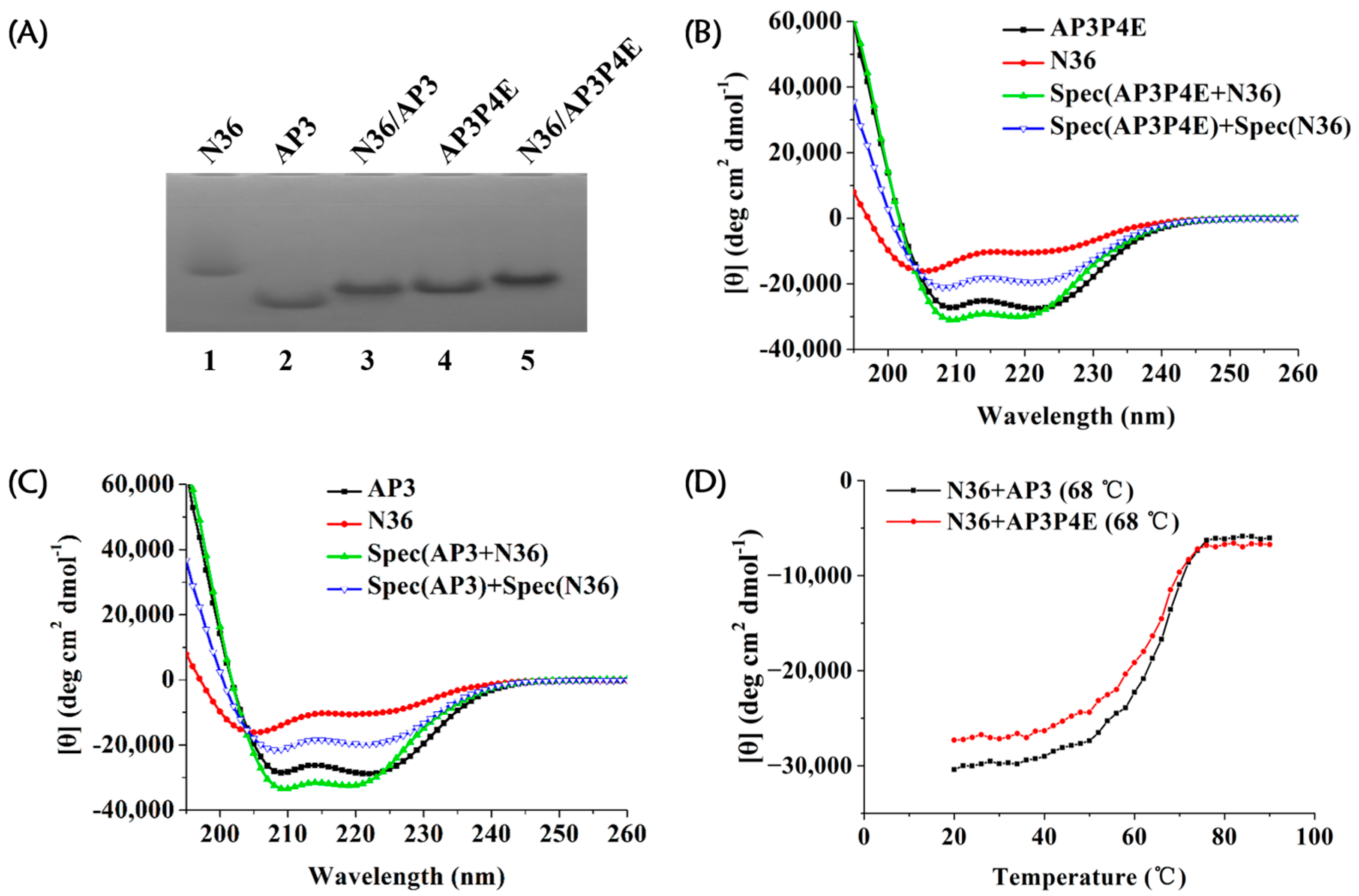
3.4. Interaction of AP3P4E with an Exogenous gp41 NHR Peptide
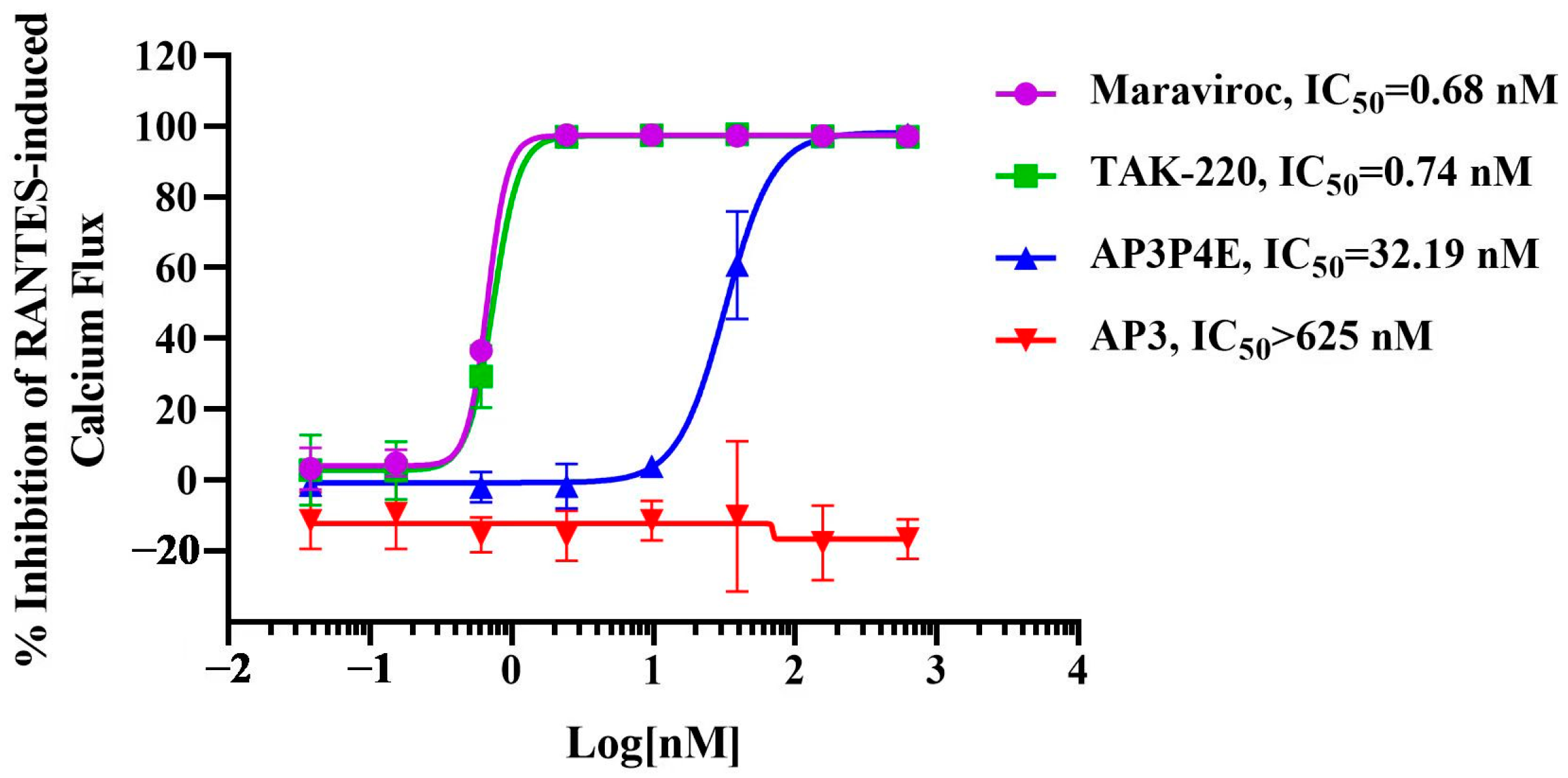
3.5. Antagonistic Activity of AP3P4E on RANTES-Induced Ca2+ Mobilization in CCR5-Expressing Cells
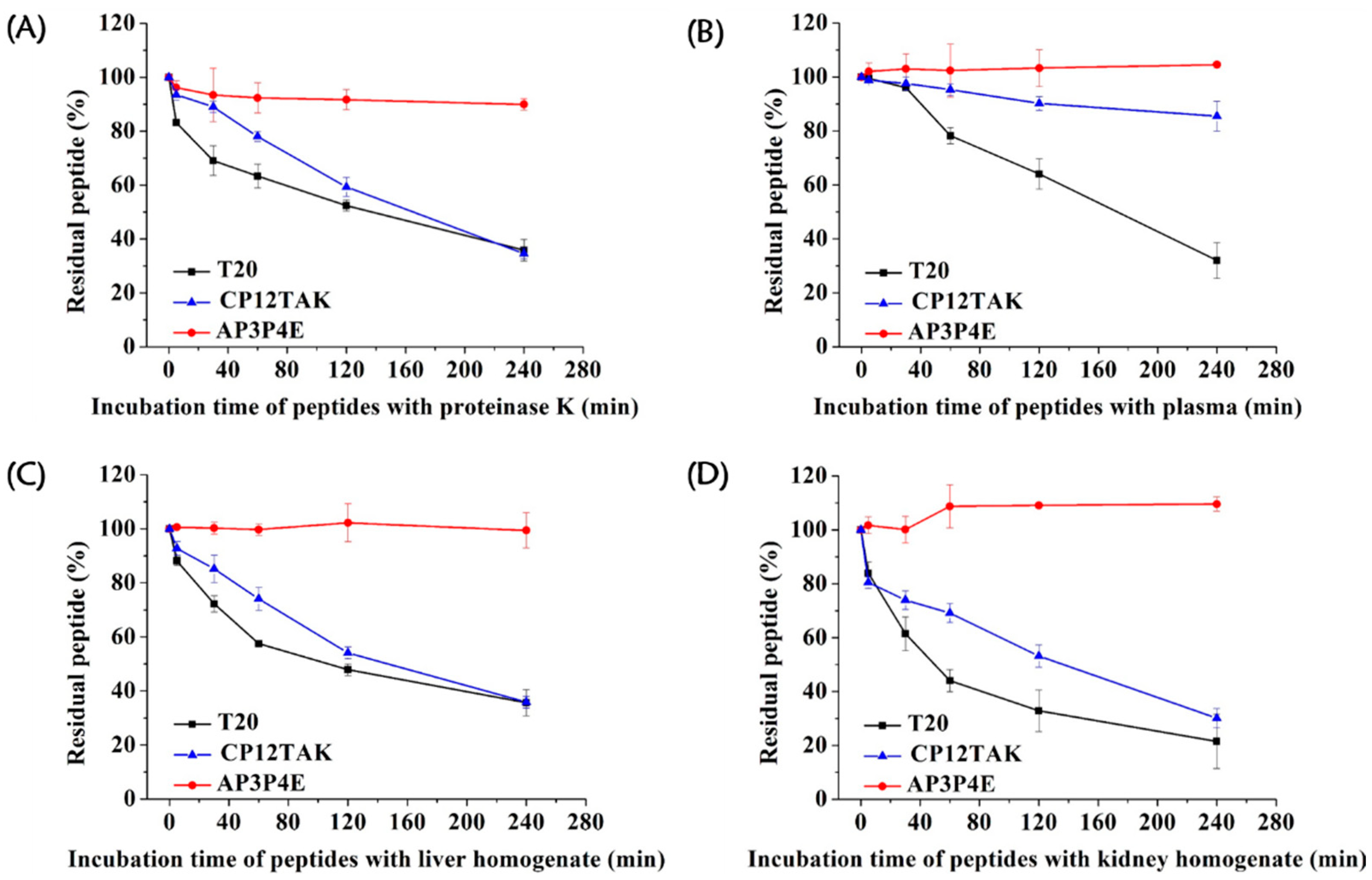
3.6. High Resistance of AP3P4E to Proteolytic Degradation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Wensing, A.M.; Calvez, V.; Ceccherini-Silberstein, F.; Charpentier, C.; Gunthard, H.F.; Paredes, R.; Shafer, R.W.; Richman, D.D. 2019 update of the drug resistance mutations in HIV-1. Top. Antivir. Med. 2019, 27, 111–121. [Google Scholar]
- Maeda, K.; Das, D.; Kobayakawa, T.; Tamamura, H.; Takeuchi, H. Discovery and Development of Anti-HIV Therapeutic Agents: Progress Towards Improved HIV Medication. Curr. Top. Med. Chem. 2019, 19, 1621–1649. [Google Scholar] [CrossRef] [PubMed]
- Menéndez-Arias, L.; Delgado, R. Update and latest advances in antiretroviral therapy. Trends Pharmacol. Sci. 2022, 43, 16–29. [Google Scholar] [CrossRef]
- Meanwell, N.A.; Krystal, M.R.; Nowicka-Sans, B.; Langley, D.R.; Conlon, D.A.; Eastgate, M.D.; Grasela, D.M.; Timmins, P.; Wang, T.; Kadow, J.F. Inhibitors of HIV-1 Attachment: The Discovery and Development of Temsavir and its Prodrug Fostemsavir. J. Med. Chem. 2018, 61, 62–80. [Google Scholar] [CrossRef]
- Tan, Q.; Zhu, Y.; Li, J.; Chen, Z.; Han, G.; Kufareva, I.; Li, T.; Ma, L.; Fenalti, G.; Li, J.; et al. Structure of the CCR5 chemokine receptor-HIV entry inhibitor maraviroc complex. Science 2013, 341, 1387–1390. [Google Scholar] [CrossRef]
- Xue, J.; Chong, H.; Zhu, Y.; Zhang, J.; Tong, L.; Lu, J.; Chen, T.; Cong, Z.; Wei, Q.; He, Y. Efficient treatment and pre-exposure prophylaxis in rhesus macaques by an HIV fusion-inhibitory lipopeptide. Cell 2022, 185, 131–144.e18. [Google Scholar] [CrossRef]
- Vigant, F.; Santos, N.C.; Lee, B. Broad-spectrum antivirals against viral fusion. Nat. Rev. Microbiol. 2015, 13, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.D.; Meng, W.; Wang, X.J.; Wang, H.C. Broad-spectrum antiviral agents. Front. Microbiol. 2015, 6, 517. [Google Scholar] [CrossRef]
- Jiang, S.; Lin, K.; Strick, N.; Neurath, A.R. HIV-1 inhibition by a peptide. Nature 1993, 365, 113. [Google Scholar] [CrossRef] [PubMed]
- Wild, C.T.; Shugars, D.C.; Greenwell, T.K.; McDanal, C.B.; Matthews, T.J. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc. Natl. Acad. Sci. USA 1994, 91, 9770–9774. [Google Scholar] [CrossRef] [PubMed]
- Furuta, R.; Wild, C.; Weng, Y.; Weiss, C. Capture of an early fusion-active conformation of HIV-1 gp41. Nat. Struct. Biol. 1998, 5, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Lalezari, J.P.; Henry, K.; O’Hearn, M.; Montaner, J.S.; Piliero, P.J.; Trottier, B.; Walmsley, S.; Cohen, C.; Kuritzkes, D.R.; Eron, J.J., Jr.; et al. Enfuvirtide, an HIV-1 fusion inhibitor, for drug-resistant HIV infection in North and South America. N. Engl. J. Med. 2003, 348, 2175–2185. [Google Scholar] [CrossRef] [PubMed]
- Lazzarin, A.; Clotet, B.; Cooper, D.; Reynes, J.; Arasteh, K.; Nelson, M.; Katlama, C.; Stellbrink, H.J.; Delfraissy, J.F.; Lange, J.; et al. Efficacy of enfuvirtide in patients infected with drug-resistant HIV-1 in Europe and Australia. N. Engl. J. Med. 2003, 348, 2186–2195. [Google Scholar] [CrossRef]
- Vincent, N.; Malvoisin, E. Ability of antibodies specific to the HIV-1 envelope glycoprotein to block the fusion inhibitor T20 in a cell-cell fusion assay. Immunobiology 2012, 217, 943–950. [Google Scholar] [CrossRef]
- Qi, Z.; Shi, W.; Xue, N.; Pan, C.; Jing, W.; Liu, K.; Jiang, S. Rationally designed anti-HIV peptides containing multifunctional domains as molecule probes for studying the mechanisms of action of the first and second generation HIV fusion inhibitors. J. Biol. Chem. 2008, 283, 30376–30384. [Google Scholar] [CrossRef]
- Shi, W.; Qi, Z.; Pan, C.; Xue, N.; Debnath, A.K.; Qie, J.; Jiang, S.; Liu, K. Novel anti-HIV peptides containing multiple copies of artificially designed heptad repeat motifs. Biochem. Biophys. Res. Commun. 2008, 374, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Cai, L.; Lu, L.; Wang, C.; Wang, K.; Xu, L.; Zhang, S.; Han, H.; Jiang, X.; Zheng, B.; et al. Design of highly potent HIV fusion inhibitors based on artificial peptide sequences. Chem. Commun. 2012, 48, 11579–11581. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, X.; Li, J.; Li, Q.; Feng, S.; Lu, L.; Wang, C.; Jiang, S. Design of artificial alpha-helical peptides targeting both gp41 deep pocket and subpocket as potent HIV-1 fusion inhibitors. Eur. J. Med. Chem. 2022, 236, 114336. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Zhu, Y.; Ye, S.; Wang, Q.; Xu, W.; Su, S.; Sun, Z.; Yu, F.; Liu, Q.; Wang, C.; et al. Improved Pharmacological and Structural Properties of HIV Fusion Inhibitor AP3 over Enfuvirtide: Highlighting Advantages of Artificial Peptide Strategy. Sci. Rep. 2015, 5, 13028. [Google Scholar] [CrossRef]
- Wang, C.; Wang, X.; Wang, H.; Pu, J.; Li, Q.; Li, J.; Liu, Y.; Lu, L.; Jiang, S. A “Two-Birds-One-Stone” Approach toward the Design of Bifunctional Human Immunodeficiency Virus Type 1 Entry Inhibitors Targeting the CCR5 Coreceptor and gp41 N-Terminal Heptad Repeat Region. J. Med. Chem. 2021, 64, 11460–11471. [Google Scholar] [CrossRef]
- Chan, D.C.; Fass, D.; Berger, J.M.; Kim, P.S. Core structure of gp41 from the HIV envelope glycoprotein. Cell 1997, 89, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Dorr, P.; Westby, M.; Dobbs, S.; Griffin, P.; Irvine, B.; Macartney, M.; Mori, J.; Rickett, G.; Smith-Burchnell, C.; Napier, C.; et al. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob. Agents Chemother. 2005, 49, 4721–4732. [Google Scholar] [CrossRef] [PubMed]
- Perry, C.M. Maraviroc: A review of its use in the management of CCR5-tropic HIV-1 infection. Drugs 2010, 70, 1189–1213. [Google Scholar] [CrossRef] [PubMed]
- Imamura, S.; Ichikawa, T.; Nishikawa, Y.; Kanzaki, N.; Takashima, K.; Niwa, S.; Iizawa, Y.; Baba, M.; Sugihara, Y. Discovery of a piperidine-4-carboxamide CCR5 antagonist (TAK-220) with highly potent Anti-HIV-1 activity. J. Med. Chem. 2006, 49, 2784–2793. [Google Scholar] [CrossRef] [PubMed]
- Akgun, E.; Javed, M.I.; Lunzer, M.M.; Powers, M.D.; Sham, Y.Y.; Watanabe, Y.; Portoghese, P.S. Inhibition of Inflammatory and Neuropathic Pain by Targeting a Mu Opioid Receptor/Chemokine Receptor5 Heteromer (MOR-CCR5). J. Med. Chem. 2015, 58, 8647–8657. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Weng, Z.; Dong, X.; Chen, L.; Ma, L.; Cen, S.; Zhou, N.; Hu, Y. Design, synthesis and biological evaluation of novel piperazine derivatives as CCR5 antagonists. PLoS ONE 2013, 8, e53636. [Google Scholar] [CrossRef] [PubMed]
- Westby, M.; Lewis, M.; Whitcomb, J.; Youle, M.; Pozniak, A.L.; James, I.T.; Jenkins, T.M.; Perros, M.; van der Ryst, E. Emergence of CXCR4-using human immunodeficiency virus type 1 (HIV-1) variants in a minority of HIV-1-infected patients following treatment with the CCR5 antagonist maraviroc is from a pretreatment CXCR4-using virus reservoir. J. Virol. 2006, 80, 4909–4920. [Google Scholar] [CrossRef] [PubMed]
- Baba, M.; Miyake, H.; Wang, X.; Okamoto, M.; Takashima, K. Isolation and characterization of human immunodeficiency virus type 1 resistant to the small-molecule CCR5 antagonist TAK-652. Antimicrob. Agents Chemother. 2007, 51, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Ray, N. Maraviroc in the treatment of HIV infection. Drug Des. Devel. Ther. 2009, 2, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Ingallinella, P.; Bianchi, E.; Ladwa, N.A.; Wang, Y.J.; Hrin, R.; Veneziano, M.; Bonelli, F.; Ketas, T.J.; Moore, J.P.; Miller, M.D.; et al. Addition of a cholesterol group to an HIV-1 peptide fusion inhibitor dramatically increases its antiviral potency. Proc. Natl. Acad. Sci. USA 2009, 106, 5801–5806. [Google Scholar] [CrossRef] [PubMed]
- Francis, J.N.; Redman, J.S.; Eckert, D.M.; Kay, M.S. Design of a modular tetrameric scaffold for the synthesis of membrane-localized D-peptide inhibitors of HIV-1 entry. Bioconjug Chem. 2012, 23, 1252–1258. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.; Wu, X.; Su, Y.; He, Y. Development of potent and long-acting HIV-1 fusion inhibitors. AIDS 2016, 30, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.; Xue, J.; Xiong, S.; Cong, Z.; Ding, X.; Zhu, Y.; Liu, Z.; Chen, T.; Feng, Y.; He, L.; et al. A Lipopeptide HIV-1/2 Fusion Inhibitor with Highly Potent In Vitro, Ex Vivo, and In Vivo Antiviral Activity. J. Virol. 2017, 91, e00288-17. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Chong, H.; Yu, D.; Guo, Y.; Zhou, Y.; He, Y. Design and Characterization of Cholesterylated Peptide HIV-1/2 Fusion Inhibitors with Extremely Potent and Long-Lasting Antiviral Activity. J. Virol. 2019, 93, e02312-18. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.; Yao, X.; Qiu, Z.; Sun, J.; Qiao, Y.; Zhang, M.; Wang, M.; Cui, S.; He, Y. The M-T hook structure increases the potency of HIV-1 fusion inhibitor sifuvirtide and overcomes drug resistance. J. Antimicrob. Chemother. 2014, 69, 2759–2769. [Google Scholar] [CrossRef] [PubMed]
- Lawless, M.; Barney, S.; Guthrie, K.; Bucy, T.; Petteway, S.; Merutka, G. HIV-1 membrane fusion mechanism: Structural studies of the interactions between biologically-active peptides from gp41. Biochemistry 1996, 35, 13697–13708. [Google Scholar] [CrossRef] [PubMed]
- Danial, M.; van Dulmen, T.H.; Aleksandrowicz, J.; Potgens, A.J.; Klok, H.A. Site-specific PEGylation of HR2 peptides: Effects of PEG conjugation position and chain length on HIV-1 membrane fusion inhibition and proteolytic degradation. Bioconjug Chem. 2012, 23, 1648–1660. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Sequence b | EC50 (nM) |
|---|---|---|
| AP3 | MTWEEWDKKIEELIKKSEELIKKIEEQIKKQEESIKK | 0.75 ± 0.22 |
| AP3W | MTWEEWDKKIEELIKKSEELIKKIEEQIKKQEESIKK(TAKW) | 6.33 ± 1.88 |
| AP3P4W | MTWEEWDKKIEELIKKSEELIKKIEEQIKKQEESIKK(PEG4-TAKW) | 0.98 ± 0.23 |
| AP3P8W | MTWEEWDKKIEELIKKSEELIKKIEEQIKKQEESIKK(PEG8-TAKW) | 0.53 ± 0.17 |
| AP3P12W | MTWEEWDKKIEELIKKSEELIKKIEEQIKKQEESIKK(PEG12-TAKW) | 2.41 ± 0.39 |
| AP3P16W | MTWEEWDKKIEELIKKSEELIKKIEEQIKKQEESIKK(PEG16-TAKW) | 4.03 ± 0.76 |
| AP3P24W | MTWEEWDKKIEELIKKSEELIKKIEEQIKKQEESIKK(PEG24-TAKW) | 8.98 ± 0.91 |
| AP3P4E | MTWEEWDKKIEELIKKSEELIKKIEEQIKKQEESIKK(PEG4-TAKE) | 0.15 ± 0.08 |
| AP3P8E | MTWEEWDKKIEELIKKSEELIKKIEEQIKKQEESIKK(PEG8-TAKE) | 0.29 ± 0.16 |
| TAK-220 | - | 23.8 ± 13.3 |
| AP3/TAK-220 c | - | 8.02 ± 3.00 |
| T20 | YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF | 4.02 ± 0.69 |
| HIV-1 Isolate | Subtype | Tropism | EC50 (nM) b | |||
|---|---|---|---|---|---|---|
| AP3 | TAK-220 | AP3P4E | T20 | |||
| 91US_4 | B | R5 | 10.7 ± 1.2 | 12.1 ± 2.7 | 3.8 ± 0.3 | 28.2 ± 2.4 |
| J32228M4 | D | R5 | 59.0 ± 6.1 | 161.0 ± 6.6 | 8.9 ± 1.4 | 59.8 ± 7.1 |
| 89BZ167 | B | X4 | 11.6 ± 0.5 | >300 | 3.09 ± 0.03 | 48.3 ± 2.9 |
| 92UG029 | A | X4 | 3.1 ± 0.1 | >300 | 1.53 ± 0.03 | 10.3 ± 2.3 |
| Compound | Solubility (mg/mL) in | |
|---|---|---|
| PBS (pH 7.4) | H2O | |
| AP3P4E | >60 | >60 |
| CP12TAK | 1.28 ± 0.04 | 1.22 ± 0.04 |
| T20 | 1.02 ± 0.03 | 0.33 ± 0.01 |
| HIV-1 Strains | EC50 (nM) b | |
|---|---|---|
| T20 | AP3P4E | |
| D36G c | 1.11 ± 0.23 | 0.97 ± 0.40 |
| N42T/N43K d | 1264.0 ± 45.1 (1138.7) | 0.17 ± 0.05 (0.2) |
| V38E/N42S d | >2000.0 (>1801.8) | 2.02 ± 0.39 (2.1) |
| V38A/N42T d | 1045.0 ± 48.6 (941.4) | 1.13 ± 0.13 (1.2) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.; Li, Q.; Sun, L.; Wang, X.; Wang, H.; Zhang, W.; Li, J.; Liu, Y.; Lu, L.; Jiang, S. An Artificial Peptide-Based Bifunctional HIV-1 Entry Inhibitor That Interferes with Viral Glycoprotein-41 Six-Helix Bundle Formation and Antagonizes CCR5 on the Host Cell Membrane. Viruses 2023, 15, 1038. https://doi.org/10.3390/v15051038
Wang C, Li Q, Sun L, Wang X, Wang H, Zhang W, Li J, Liu Y, Lu L, Jiang S. An Artificial Peptide-Based Bifunctional HIV-1 Entry Inhibitor That Interferes with Viral Glycoprotein-41 Six-Helix Bundle Formation and Antagonizes CCR5 on the Host Cell Membrane. Viruses. 2023; 15(5):1038. https://doi.org/10.3390/v15051038
Chicago/Turabian StyleWang, Chao, Qing Li, Lujia Sun, Xinling Wang, Huan Wang, Wenpeng Zhang, Jiahui Li, Yang Liu, Lu Lu, and Shibo Jiang. 2023. "An Artificial Peptide-Based Bifunctional HIV-1 Entry Inhibitor That Interferes with Viral Glycoprotein-41 Six-Helix Bundle Formation and Antagonizes CCR5 on the Host Cell Membrane" Viruses 15, no. 5: 1038. https://doi.org/10.3390/v15051038
APA StyleWang, C., Li, Q., Sun, L., Wang, X., Wang, H., Zhang, W., Li, J., Liu, Y., Lu, L., & Jiang, S. (2023). An Artificial Peptide-Based Bifunctional HIV-1 Entry Inhibitor That Interferes with Viral Glycoprotein-41 Six-Helix Bundle Formation and Antagonizes CCR5 on the Host Cell Membrane. Viruses, 15(5), 1038. https://doi.org/10.3390/v15051038