
A Multiplexed, Tiled PCR Method for Rapid Whole-Genome Sequencing of Infectious Spleen and Kidney Necrosis Virus (ISKNV) in Tilapia

 , , , ,
, , , ,
Abstract
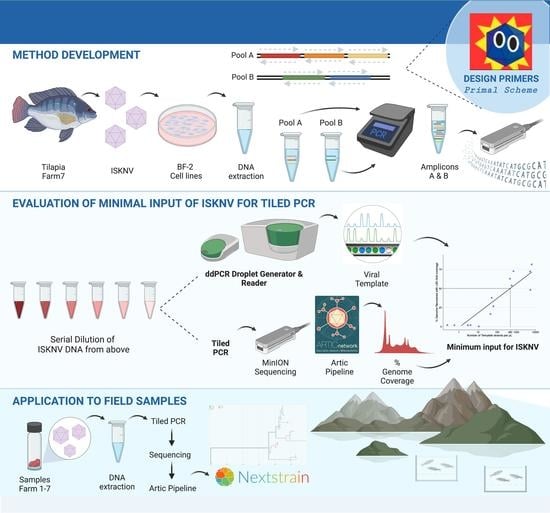
1. Introduction
2. Materials and Methods
2.1. Samples and DNA Extraction
2.2. Design of Primers
2.3. Primer Pool Preparation
2.4. Failed Regions Recovered Using Neighbouring Pairs
2.5. Library Preparation, Sequencing Protocols and Bioinformatic Processing
2.6. Construction of the Full ISKNV Genome Infecting Tilapia in Ghana
2.7. Droplet Digital PCR to Determine Minimal Input for Genome Recovery of ISKNV Using the Tiled PCR Protocol
2.8. Epidemiology and Phylogeographic Analysis of ISKNV
2.9. Tiled PCR for MinION Sequencing of ISKNV Directly from Samples Collected from Lake Volta Outbreak
3. Results
3.1. Tiled PCR Recovers near Complete ISKNV Genome from Cell-Line Extracts
3.2. ddPCR Determined Minimal Input for Genome Recovery of ISKNV Using the Tiled PCR Protocol
3.3. Epidemiology and Phylogeographic Analysis of ISKNV Is Not Solely Related to Host Species
3.4. Phylogenetic Analysis Indicates Multiple Introductions of ISKNV in Fish Samples Collected from Lake Volta
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- FAO. 2020. Available online: http://www.fao.org/documents/card/en/c/ca9229en (accessed on 10 February 2023).
- Machimbirike, V.I.; Jansen, M.D.; Senapin, S.; Khunrae, P.; Rattanarojpong, T.; Dong, H.T. Viral infections in tilapines: More than just tilapia lake virus. Aquaculture 2019, 503, 508–518. [Google Scholar] [CrossRef]
- Eyngor, M.; Zamostiano, R.; Tsofack, J.E.K.; Berkowitz, A.; Bercovier, H.; Tinman, S.; Lev, M.; Hurvitz, A.; Galeotti, M.; Bacharach, E.; et al. Identification of a novel RNA virus lethal to tilapia. J. Clin. Microbiol. 2014, 52, 4137–4146. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Lu, M. Tilapia polyculture: A global review. Aquac. Res. 2016, 47, 2363–2374. [Google Scholar] [CrossRef]
- Sasu, D.D. Ghana: Annual Volume of Tilapia Production. Statista. 2021. Available online: https://www.statista.com/statistics/1278956/production-volume-of-tilapia-in-ghana/ (accessed on 3 March 2023).
- Verner-Jeffreys, D.W.; Wallis, T.J.; Cejas, I.C.; Ryder, D.; Haydon, D.J.; Domazoro, J.F.; Dontwi, J.; Field, T.R.; Adjei-Boteng, D.; Wood, G.; et al. Streptococcus agalactiae Multilocus sequence type 261 is associated with mortalities in the emerging Ghanaian tilapia industry. J. Fish Dis. 2018, 41, 175–179. [Google Scholar] [CrossRef] [PubMed]
- McMurtrie, J.; Alathari, S.; Chaput, D.L.; Bass, D.; Ghambi, C.; Nagoli, J.; Delamare-Deboutteville, J.; Mohan, C.V.; Cable, J.; Temperton, B.; et al. Relationships between pond water and tilapia skin microbiomes in aquaculture ponds in Malawi. Aquaculture. 2022, 558, 73836710. [Google Scholar] [CrossRef]
- Sivasankar, P. A Review on DNA Viral Diseases of Fish. 2017. Available online: https://www.fisheriesjournal.com/archives/2017/vol5issue4/PartF/5-4-44-714.pdf (accessed on 11 February 2023).
- Dong, H.T.; Nguyen, V.V.; Le, H.D.; Sangsuriya, P.; Jitrakorn, S.; Saksmerprome, V.; Senapin, S.; Rodkhum, C. Naturally concurrent infections of bacterial and viral pathogens in disease outbreaks in cultured Nile tilapia (Oreochromis niloticus) farms. Aquaculture 2015, 448, 427–435. [Google Scholar] [CrossRef]
- Dong, H.; Siriroob, S.; Meemetta, W.; Santimanawong, W.; Gangnonngiw, W.; Pirarat, N.; Khunrae, P.; Rattanarojpong, T.; Vanichviriyakit, R.; Senapin, S. Emergence of tilapia lake virus in Thailand and an alternative semi-nested RT-PCR for detection. Aquaculture 2017, 476, 111–118. [Google Scholar] [CrossRef]
- Ramírez-Paredez, J.G.; Paley, R.K.; Hunt, W.; Feist, S.W.; Stone, D.M.; Field, T.R.; Haydon, D.J.; Ziddah, P.A.; Nkansa, M.; Guilder, J.; et al. First detection of Infectious Spleen and kidney Necrosis Virus (ISKNV) associated with massive mortalities in farmed tilapia in Africa. Transbound. Emerg. Dis. 2021, 68, 680538. [Google Scholar] [CrossRef]
- Mohr, P.; Moody, N.; Williams, L.; Hoad, J.; Cummins, D.; Davies, K.; Crane, M.S. Molecular confirmation of infectious spleen and kidney necrosis virus (ISKNV) in farmed and imported ornamental fish in Australia. Dis. Aquat. Organ. 2015, 116, 103–110. [Google Scholar] [CrossRef]
- Fu, X.; Li, N.; Liu, L.; Lin, Q.; Wang, F.; Lai, Y.; Jiang, H.; Pan, H.; Shi, C.; Wu, S. Genotype and host range analysis of infectious spleen and kidney necrosis virus (ISKNV). Virus Genes 2011, 42, 97–109. [Google Scholar] [CrossRef]
- He, J.G.; Denga, M.; Weng, S.P.; Lia, Z.; Zhou, S.Y.; Long, Q.X.; Wang, X.Z.; Chanb, S.M. Complete genome analysis of the mandarin fish infectious spleen and kidney necrosis iridovirus. Virology 2001, 291, 126–139. [Google Scholar] [CrossRef] [PubMed]
- He, J.G.; Zeng, K.; Weng, S.P.; Chan, S.M. Experimental transmission, pathogenicity and physical–chemical properties of infectious spleen and kidney necrosis virus (ISKNV). Aquaculture 2002, 204, 11–24. [Google Scholar] [CrossRef]
- Jung-Schroers, V.; Adamek, M.; Wohlsein, P.; Wolter, J.; Wedekind, H.; Steinhagen, D. First outbreak of an infection with infectious spleen and kidney necrosis virus (ISKNV) in ornamental fish in Germany. Dis. Aquat. Organ. 2016, 119, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Howell, M. Seven Deadly Fins: Study Sheds Light on Key Tilapia Viruses [Internet]. 2019. Available online: https://thefishsite.com/articles/seven-deadly-fins-study-sheds-light-on-key-tilapia-viruses (accessed on 5 December 2019).
- Assefa, A.; Abunna, F. Maintenance of Fish Health in Aquaculture: Review of Epidemiological Approaches for Prevention and Control of Infectious Disease of Fish. Vet. Med. Int. 2018, 2018, 5432497. [Google Scholar] [CrossRef]
- Avarre, J.C. Editorial: Molecular Tracing of Aquatic Viruses: Where Epidemiology Needs to Meet Genomics. Front. Microbiol. 2017, 8, 1498. [Google Scholar] [CrossRef]
- Gilchrist, C.A.; Turner, S.D.; Riley, M.F.; Petri, W.A., Jr.; Hewlett, E.L. Whole-genome sequencing in outbreak analysis. Clin. Microbiol. Rev. 2015, 28, 541–563. [Google Scholar] [CrossRef]
- Quick, J.; Zivin, K.; Turiczek, J.; Sherkow, J.S.; Lane, M.; Skerrett, P. Real-Time GENE sequencing Can Help Control Pandemics. 2020. Available online: https://www.statnews.com/2020/09/11/real-time-gene-sequencing-can-help-control-and-may-someday-prevent-pandemics/ (accessed on 1 February 2023).
- Quick, J.; Loman, N.J.; Duraffour, S.; Simpson, J.T.; Severi, E.; Cowley, L.; Bore, J.A.; Koundouno, R.; Dudas, G.; Mikhail, A.; et al. Real-time, portable genome sequencing for Ebola surveillance. Nature 2016, 530, 228–232. [Google Scholar] [CrossRef]
- Quick, J.; Grubaugh, N.D.; Pullan, S.T.; Claro, I.M.; Smith, A.D.; Gangavarapu, K.; Oliveira, G.; Robles-Sikisaka, R.; Rogers, T.F.; Beutler, A.N.; et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 2017, 12, 1261–1276. [Google Scholar] [CrossRef]
- Tyson, J.R.; James, P.; Stoddart, D.; Sparks, N.; Wickenhagen, A.; Hall, G.; Choi, J.H.; Lapointe, H.; Kamelian, K.; Smith, A.D.; et al. Improvements to the ARTIC multiplex PCR method for SARS-CoV-2 genome sequencing using nanopore. bioRxiv 2020. [Google Scholar] [CrossRef]
- Resende, P.C.; Motta, F.C.; Roy, S.; Appolinario, L.; Fabri, A.; Xavier, J.; Harris, K.; Matos, A.R.; Caetano, B.; Orgeswalska, M.; et al. SARS-CoV-2 Genomes Recovered by Long Amplicon Tiling Multiplex Approach Using Nanopore Sequencing and Applicable to Other Sequencing Platforms. Cold Spring Harbor Laboratory. 2020. Available online: https://www.biorxiv.org/content/10.1101/2020.04.30.069039v1 (accessed on 23 January 2021).
- Du Plessis, L.; McCrone, J.T.; Zarebski, A.E.; Hill, V.; Ruis, C.; Gutierrez, B.; Raghwani, J.; Ashworth, J.; Colquhoun, R.; Connor, T.R.; et al. Establishment and lineage dynamics of the SARS-CoV-2 epidemic in the UK. Science 2021, 371, 708–712. [Google Scholar] [CrossRef]
- Jiang, N.; Shen, J.; Zhou, Y.; Liu, W.; Meng, Y.; Li, Y.; Xue, M.; Xu, C.; Fan, Y. Development of a droplet digital PCR method for the sensitive detection and quantification of largemouth bass ranavirus. J. Fish Dis. 2023, 46, 91–98. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Milne, I.; Stephen, G.; Bayer, M.; Cock, P.; Pritchard, L.; Cardle, L.; Shaw, P.D.; Marshall, D. Using Tablet for visual exploration of second-generation sequencing data. Brief. Bioinform. 2013, 14, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Fu, X.; Liu, L.; Liang, H.; Guo, H.; Yin, S.; Kumaresan, V.; Huang, Z.; Li, N. Application and development of a TaqMan real-time PCR for detecting infectious spleen and kidney necrosis virus in Siniperca chuatsi. Microb. Pathog. 2017, 107, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.I.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Wang, Y.Q.; Lü, L.; Weng, S.P.; Huang, J.N.; Chan, S.M.; He, J.G. Molecular epidemiology and phylogenetic analysis of a marine fish infectious spleen and kidney necrosis virus-like (ISKNV-like) virus. Arch. Virol. 2007, 152, 763–773. [Google Scholar] [CrossRef]
- Andrei, G.; Balzarini, J.; Fiten, P.; De Clercq, E.; Opdenakker, G.; Snoeck, R. Characterization of herpes simplex virus type 1 thymidine kinase mutants selected under a single round of high-dose brivudin. J. Virol. 2005, 79, 5863–5869. [Google Scholar] [CrossRef]
- Xie, Y.; Wu, L.; Wang, M.; Cheng, A.; Yang, Q.; Wu, Y.; Jia, R.; Zhu, D.; Zhao, X.; Chen, S.; et al. Alpha-Herpesvirus Thymidine Kinase Genes Mediate Viral Virulence and Are Potential Therapeutic Targets. Front. Microbiol. 2019, 10, 941. [Google Scholar] [CrossRef] [PubMed]
- Odon, V.; Georgana, I.; Holley, J.; Morata, J.; Maluquer de Motes, C. Novel Class of Viral Ankyrin Proteins Targeting the Host E3 Ubiquitin Ligase Cullin-2. J. Virol. 2018, 92, e01374-18. [Google Scholar] [CrossRef] [PubMed]
- Zeng, R.; Pan, W.; Lin, Y.; He, J.; Luo, Z.; Li, Z.; Weng, S.; He, J.; Guo, C. Development of a gene-deleted live attenuated candidate vaccine against fish virus (ISKNV) with low pathogenicity and high protection. iScience 2021, 24, 102750. [Google Scholar] [CrossRef] [PubMed]
- Lambisia, A.W.; Mohammed, K.S.; Makori, T.O.; Ndwiga, L.; Mburu, M.W.; Morobe, J.M.; Moraa, E.O.; Musyoki, J.; Murunga, N.; Mwangi, J.N.; et al. Optimization of the SARS-CoV-2 ARTIC Network V4 Primers and Whole Genome Sequencing Protocol. Front. Med. 2022, 9, 836728. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.W. The distribution of rates of spontaneous mutation over viruses, prokaryotes, and eukaryotes. Ann. N. Y. Acad. Sci. 1999, 870, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Eigen, M.; Schuster, P. The hypercycle. A principle of natural self-organization. Part A: Emergence of the hypercycle. Naturwissenschaften 1977, 64, 541–565. [Google Scholar] [CrossRef]
- Grubaugh, N.D.; Gangavarapu, K.; Quick, J.; Matteson, N.L.; De Jesus, J.G.; Main, B.J.; Tan, A.L.; Paul, L.M.; Brackney, D.E.; Grewal, S.; et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019, 20, 8. [Google Scholar] [CrossRef]
- Kerddee, P.; Dinh-Hung, N.; Dong, H.T.; Hirono, I.; Soontara, C.; Areechon, N.; Srisapo, P.; Kayansamruaj, P. Molecular evidence for homologous strains of infection spleen and kidney necrosis virus(ISKNV) genotype I infecting inland freshwater cultured Asian sea bass (Lates calcarifer) in Thailand. Arch. Virol. 2021, 166, 3061–3074. [Google Scholar] [CrossRef]
- Koda, S.A.; Subramaniam, K.; Pouder, B.; Yanong Roy, P.; Frasca, J.r.S.; Popov, V.L.; Waltzek, T.B. Complete genome sequences of infectious spleen and kidney necrosis virus isolated from farmed albino rainbow sharks Epalzeorhynchos frenatum in the United States. Virus Genes 2021, 57, 448–452. [Google Scholar] [CrossRef]
- Fusianto, C.; Hick, P.M.; Murwantoko, H.A.; Herlambang, A.; Whittington, R.J.; Becker, A.J. Outbreak investigation attributes Infectious spleen and kidney necrosis virus as a necessary cause of a mortality epidemic in farmed grouper (Epinephelus spp.) in Bali, Indonesia. Aquac. Rep. 2021, 20, 100723. [Google Scholar] [CrossRef]
- Figueiredo, H.C.P.; Tavares, G.C.; Dorella, F.A.; Rosa, J.C.C.; Marcelino, S.A.C.; Pierezan, F.; Pereira, F.L. First report of infectious spleen and kidney necrosis virus in Nile tilapia in Brazil. Transbound. Emerg. Dis. 2022, 69, 3008–3015. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Farm | Number of Samples | Date | Region |
|---|---|---|---|
| 1 | 5 | 18 October 2018 | Dodi |
| 2 | 11 | 28 November 2018 | Asikuma |
| 2 | 5 | 20 May 2022 | Asikuma |
| 6 | 10 | 10 July 2019 | Dasasi |
| 7 | 5 | 11 July 2019 | Akosombo |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alathari, S.; Chaput, D.L.; Bolaños, L.M.; Joseph, A.; Jackson, V.L.N.; Verner-Jeffreys, D.; Paley, R.; Tyler, C.R.; Temperton, B. A Multiplexed, Tiled PCR Method for Rapid Whole-Genome Sequencing of Infectious Spleen and Kidney Necrosis Virus (ISKNV) in Tilapia. Viruses 2023, 15, 965. https://doi.org/10.3390/v15040965
Alathari S, Chaput DL, Bolaños LM, Joseph A, Jackson VLN, Verner-Jeffreys D, Paley R, Tyler CR, Temperton B. A Multiplexed, Tiled PCR Method for Rapid Whole-Genome Sequencing of Infectious Spleen and Kidney Necrosis Virus (ISKNV) in Tilapia. Viruses. 2023; 15(4):965. https://doi.org/10.3390/v15040965
Chicago/Turabian StyleAlathari, Shayma, Dominique L. Chaput, Luis M. Bolaños, Andrew Joseph, Victoria L. N. Jackson, David Verner-Jeffreys, Richard Paley, Charles R. Tyler, and Ben Temperton. 2023. "A Multiplexed, Tiled PCR Method for Rapid Whole-Genome Sequencing of Infectious Spleen and Kidney Necrosis Virus (ISKNV) in Tilapia" Viruses 15, no. 4: 965. https://doi.org/10.3390/v15040965
APA StyleAlathari, S., Chaput, D. L., Bolaños, L. M., Joseph, A., Jackson, V. L. N., Verner-Jeffreys, D., Paley, R., Tyler, C. R., & Temperton, B. (2023). A Multiplexed, Tiled PCR Method for Rapid Whole-Genome Sequencing of Infectious Spleen and Kidney Necrosis Virus (ISKNV) in Tilapia. Viruses, 15(4), 965. https://doi.org/10.3390/v15040965