

Metagenomic Detection of Divergent Insect- and Bat-Associated Viruses in Plasma from Two African Individuals Enrolled in Blood-Borne Surveillance

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Specimen Sourcing
2.2. Nucleic Acid Extraction
2.3. DNA Library Synthesis
2.4. Next-Generation Sequencing
2.5. Genome Assembly
2.6. Minor Variant Analysis
2.7. Phylogenetic Analysis
2.8. Putative Host Assignment
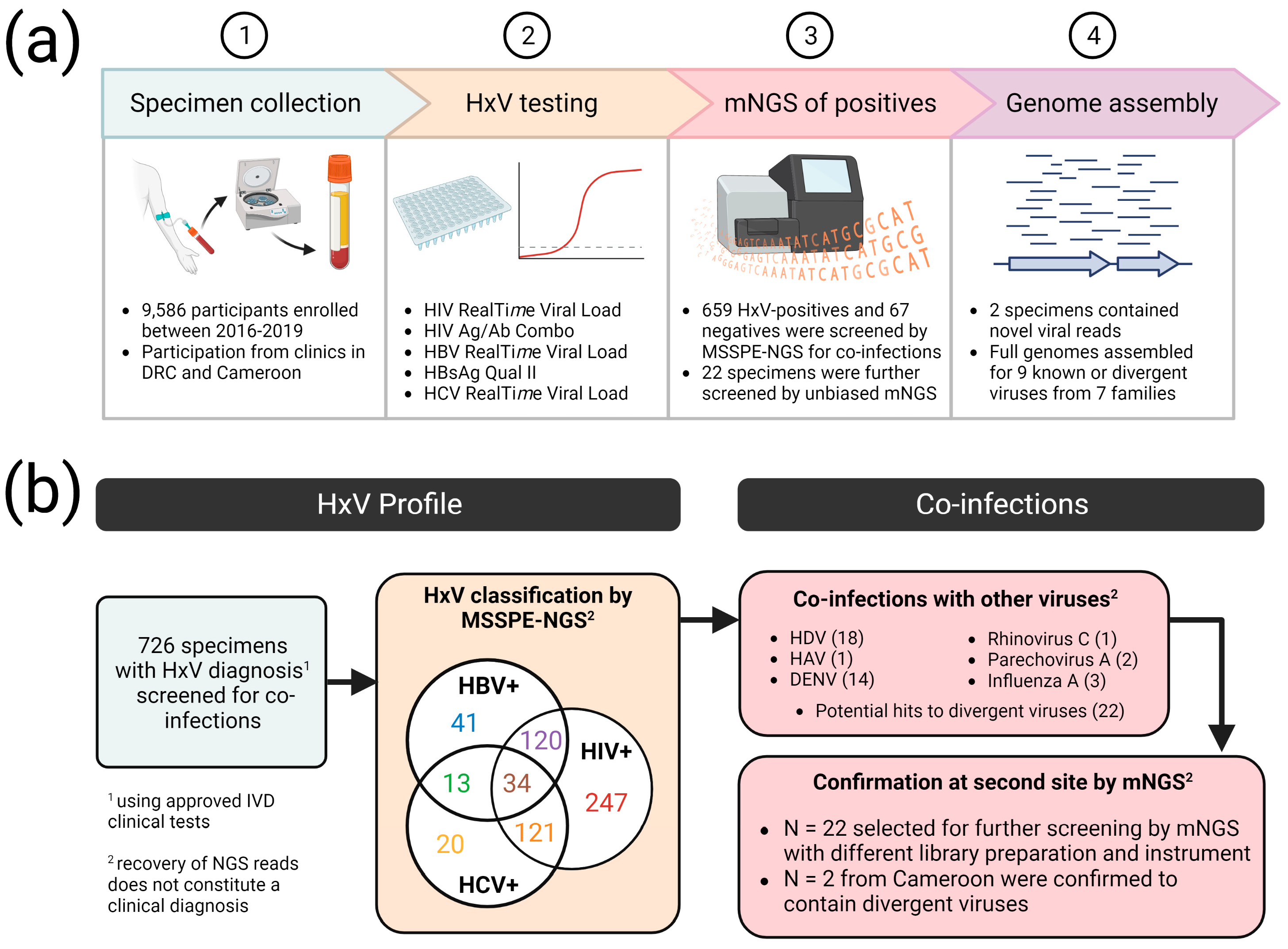
3. Results
3.1. Identification of Multiple Viruses in HxV-Positive Specimens
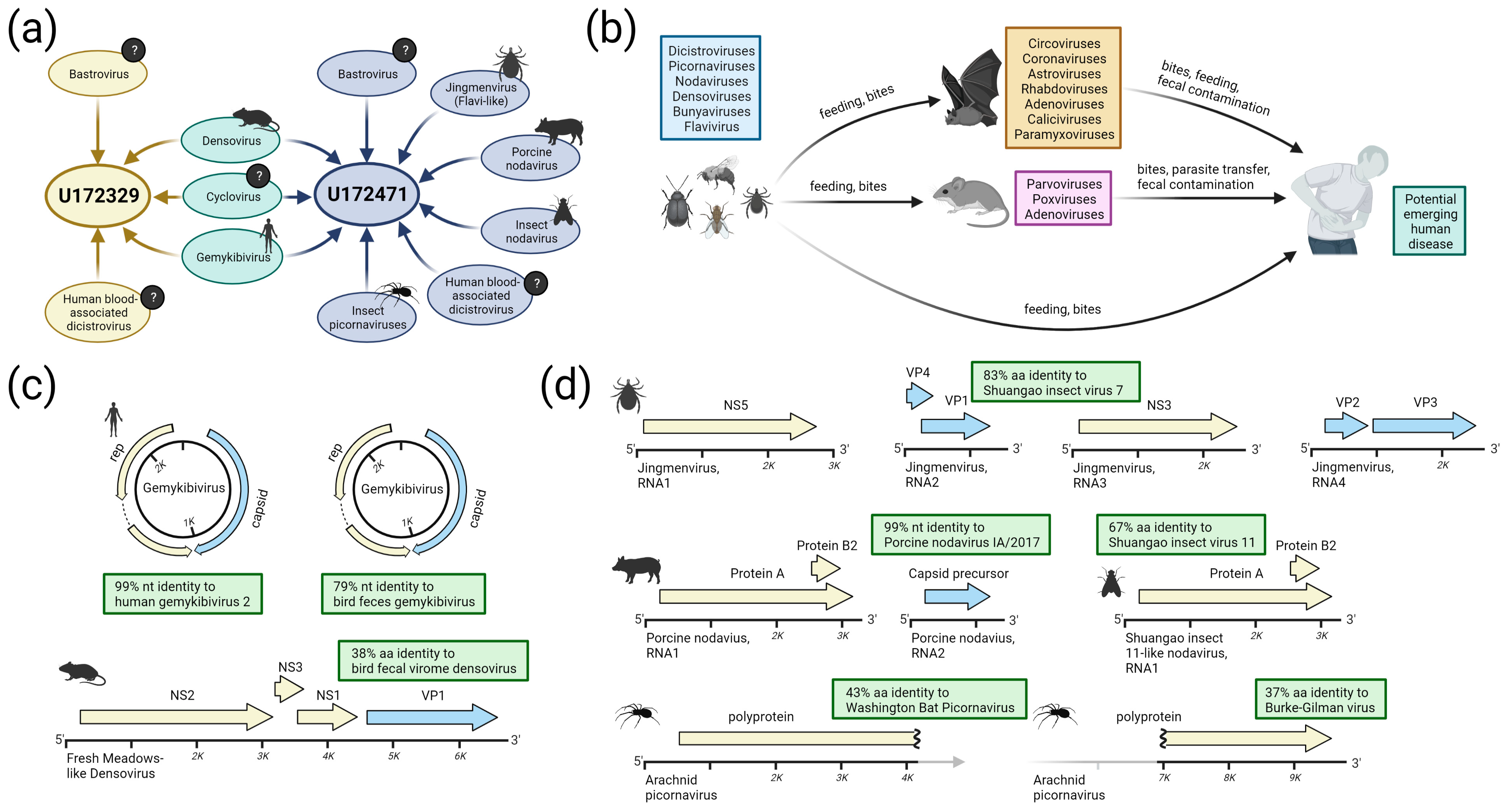
3.2. Detection of Known and Divergent Insect-Related Viruses
3.2.1. Gemykibiviruses
3.2.2. Flavi-like Viruses
3.2.3. Densoviruses
3.2.4. Nodaviruses
3.2.5. Picornaviruses
3.3. Detection of Viruses with Potential for Vertebrate Infection
3.3.1. Dicistroviruses
3.3.2. Cycloviruses
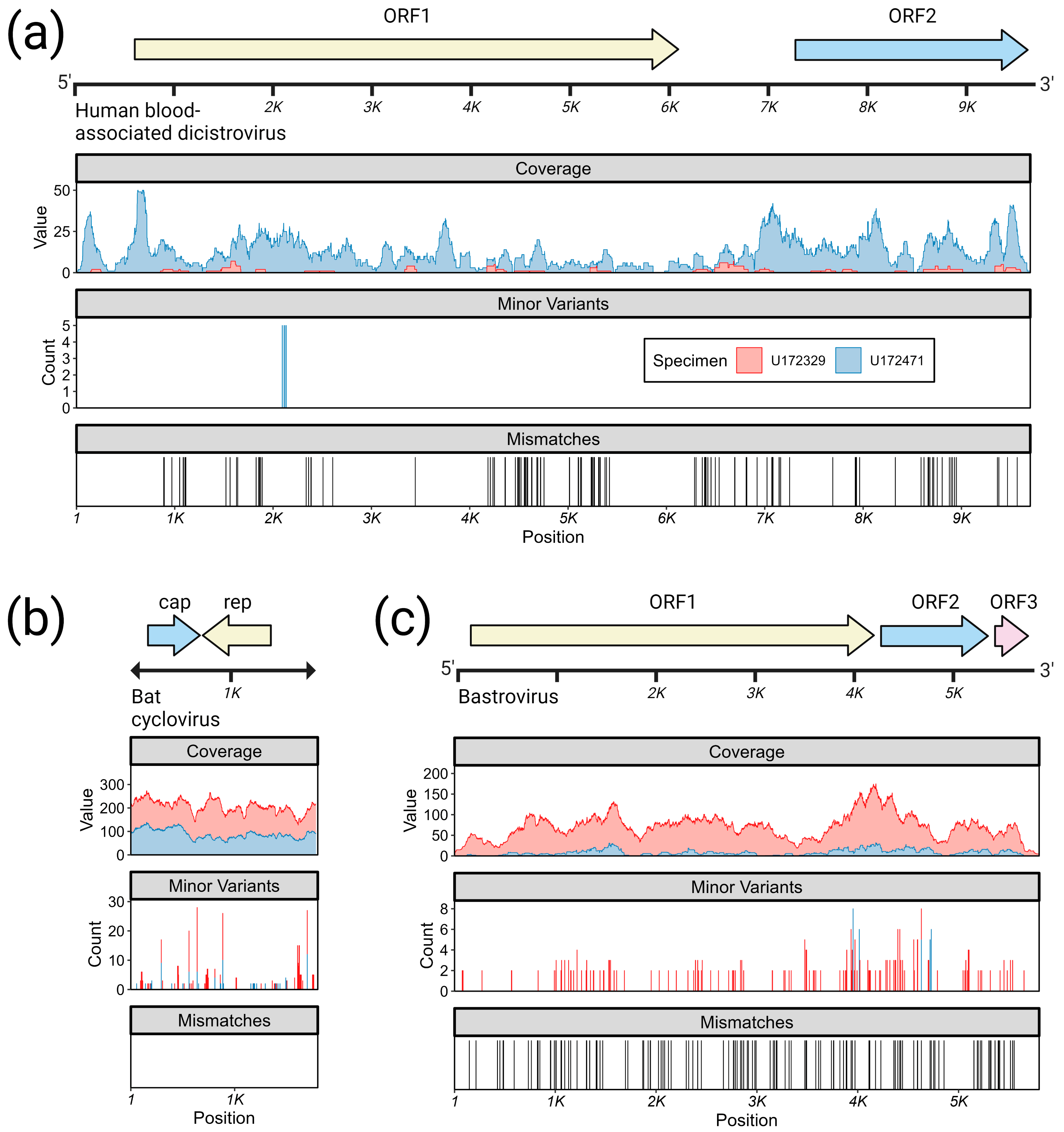
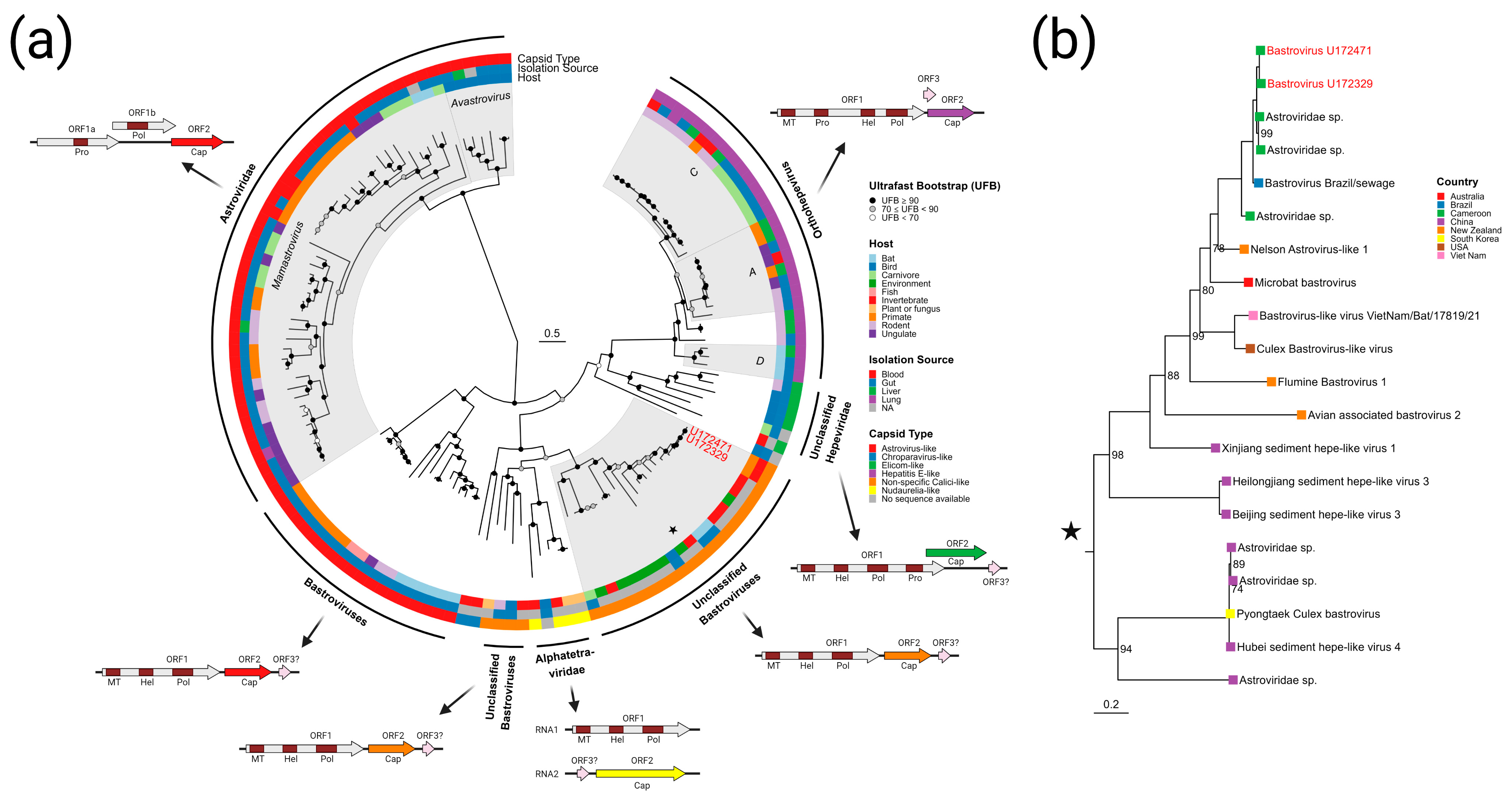
3.3.3. Bastroviruses
3.4. Evaluation of Possible Host Range
4. Discussion
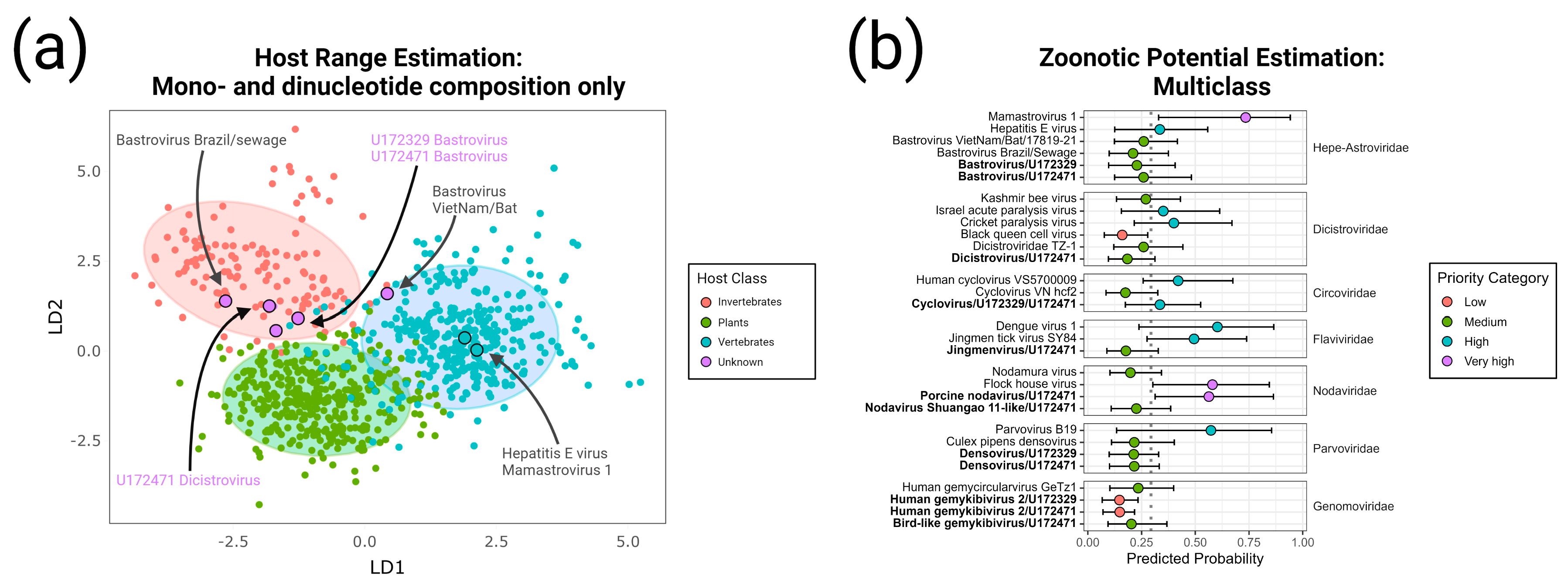
- Bastrovirus: In describing the initial discovery of bastroviruses in human stool, Oude Munnink et al. [63] suggested that sustained PCR detection over decades and accumulated genetic diversity in the capsid proteins showed the viruses had been circulating in humans or another host for some time. Bastroviruses are also prevalent globally, with recent detection in North America [82], South America [83], Asia [84], Oceania [85], and Africa [72]. Notably, the most closely related isolates to our presented bastroviruses were found in Cameroonian shellfish [86], sampled within 200 km of where our human subjects were located. As the genomic sequences are 97% identical at the amino acid level, it is possible that these viruses have been cryptically circulating in the shellfish reservoir or human population. We observed a similar profile of viral families (e.g., cyclovirus, densovirus, picornavirus) to that observed in that same metagenomic survey [86], but also in North American shellfish [87]. These lines of evidence point to shellfish consumption being a possible source for human infections from several of the detected viruses (if these viruses are indeed infectious). Of note, no clinical disease symptoms have been statistically associated with the presence of bastrovirus [63]. However, the relatedness of bastroviruses to established human pathogens such as astroviruses and hepatitis E virus, both of which are transmitted by contaminated food or water, warrants further attention. Based on the phylogenetic analysis presented in Figure 4, the acquisition of new capsid types and new ORF3s are the likely drivers of host-jumping events (indeed, most minor variants detected in our bastroviruses appeared in the capsid, see Figure 3c). This has likely happened before with the hepatitis E-like viruses, with an ancestral invertebrate- or bird-infecting virus jumping to rodents and a later descendent jumping to bats and primates (Figure 4). More work is needed to re-classify bastroviruses and better explore the true host ranges of the various clades, especially since some members seem to be adapting vertebrate-like genomic nucleotide compositions (Figure 5).
- Dicistrovirus: While dicistroviruses are believed to exclusively infect invertebrates, they have been detected in patients with febrile illness in Peru and Tanzania [58,59]. Two different dicistroviruses were detected in U172329 and U172471 with >90% nucleotide identity to the virus found in the Peruvian patient population. Only one position in the genome from U172471 had a minor variant, suggesting that these viruses may either be contaminants or may not be replicating in the primary (e.g., insect) or incidental (e.g., human) host. Nonetheless, the zoonotic potential analysis in Figure 5 suggests potential human infectivity in other dicistroviruses, so further investigation of this virus family is warranted.
- Cyclovirus: Both U172329 and U17471 possessed an identical cyclovirus genome with most minor variants detected in capsid or the intergenic, untranslated region. This may indicate immune evasion and lack of selective pressure in the natural host, respectively. Our sequence’s closest relatives have been found in mongoose feces [61], human feces [88], human respiratory tracts [89], rodents feces [78], bat feces [72], winged insects [90], and chicken muscle [91]. Moderate identity is seen with cycloviruses isolated from human plasma, respiratory tract, and CSF, with and without associated clinical manifestations such as encephalitis, respiratory illness, and sepsis [53,62,92,93,94]. Since disparate groups of cycloviruses continue to be discovered in human specimens, we share some concern that this viral family may contain members capable of zoonotic disease. Indeed, it has been suggested that dietary and environmental sources of exposure lead to unexpected new ecological niches for small DNA viruses such as cycloviruses [95].
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wolfe, N.D.; Dunavan, C.P.; Diamond, J. Origins of major human infectious diseases. Nature 2007, 447, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Grubaugh, N.D.; Ladner, J.T.; Lemey, P.; Pybus, O.G.; Rambaut, A.; Holmes, E.C.; Andersen, K.G. Tracking virus outbreaks in the twenty-first century. Nat. Microbiol. 2019, 4, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Plowright, R.K.; Parrish, C.R.; Mccallum, H.; Hudson, P.J.; Ko, A.I.; Graham, A.L.; Lloyd-Smith, J.O. Pathways to zoonotic spillover. Nat. Rev. Microbiol. 2017, 15, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.F.; Crameri, G. Emerging zoonotic viral diseases. Rev. Sci. Tech. 2014, 33, 569–581. [Google Scholar] [CrossRef]
- Peiris, J.; Lai, S.; Poon, L.; Guan, Y.; Yam, L.; Lim, W.; Nicholls, J.; Yee, W.; Yan, W.; Cheung, M.; et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 2003, 361, 1319–1325. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Sharp, P.M.; Hahn, B.H. Origins of HIV and the AIDS Pandemic. Cold Spring Harb. Perspect. Med. 2011, 1, a006841. [Google Scholar] [CrossRef]
- Colpitts, T.M.; Conway, M.J.; Montgomery, R.R.; Fikrig, E. West Nile Virus: Biology, Transmission, and Human Infection. Clin. Microbiol. Rev. 2012, 25, 635–648. [Google Scholar] [CrossRef]
- Hanley, K.A.; Monath, T.P.; Weaver, S.C.; Rossi, S.L.; Richman, R.L.; Vasilakis, N. Fever versus fever: The role of host and vector susceptibility and interspecific competition in shaping the current and future distributions of the sylvatic cycles of dengue virus and yellow fever virus. Infect. Genet. Evol. 2013, 19, 292–311. [Google Scholar] [CrossRef]
- Epstein, J.H.; Anthony, S.J.; Islam, A.; Kilpatrick, A.M.; Ali Khan, S.; Balkey, M.D.; Ross, N.; Smith, I.; Zambrana-Torrelio, C.; Tao, Y.; et al. Nipah virus dynamics in bats and implications for spillover to humans. Proc. Natl. Acad. Sci. USA 2020, 117, 29190–29201. [Google Scholar] [CrossRef]
- Gurley, E.S.; Hegde, S.T.; Hossain, K.; Sazzad, H.M.S.; Hossain, M.J.; Rahman, M.; Sharker, M.A.Y.; Salje, H.; Islam, M.S.; Epstein, J.H.; et al. Convergence of Humans, Bats, Trees, and Culture in Nipah Virus Transmission, Bangladesh. Emerg. Infect. Dis. 2017, 23, 1446–1453. [Google Scholar] [CrossRef] [PubMed]
- Kemp, S.A.; Collier, D.A.; Datir, R.P.; Ferreira, I.; Gayed, S.; Jahun, A.; Hosmillo, M.; Rees-Spear, C.; Mlcochova, P.; Lumb, I.U.; et al. SARS-CoV-2 evolution during treatment of chronic infection. Nature 2021, 592, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Vanichanan, J.; Udomkarnjananun, S.; Avihingsanon, Y.; Jutivorakool, K. Common viral infections in kidney transplant recipients. Kidney Res. Clin. Pract. 2018, 37, 323–337. [Google Scholar] [CrossRef]
- Pandey, A.; Galvani, A.P. The global burden of HIV and prospects for control. Lancet HIV 2019, 6, e809–e811. [Google Scholar] [CrossRef] [PubMed]
- Sheena, B.S.; Hiebert, L.; Han, H.; Ippolito, H.; Abbasi-Kangevari, M.; Abbasi-Kangevari, Z.; Abbastabar, H.; Abdoli, A.; Abubaker Ali, H.; Adane, M.M.; et al. Global, regional, and national burden of hepatitis B, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet Gastroenterol. Hepatol. 2022, 7, 796–829. [Google Scholar] [CrossRef] [PubMed]
- Brunner, N.; Bruggmann, P. Trends of the Global Hepatitis C Disease Burden: Strategies to Achieve Elimination. J. Prev. Med. Public Health 2021, 54, 251–258. [Google Scholar] [CrossRef]
- Muehlenbein, M.P. Human-Wildlife Contact and Emerging Infectious Diseases. In Human-Environment Interactions; Springer: Dordrecht, The Netherlands, 2013; pp. 79–94. [Google Scholar]
- Ellwanger, J.H.; Chies, J.A.B. Zoonotic spillover: Understanding basic aspects for better prevention. Genet. Mol. Biol. 2021, 44 (Suppl. 1), e20200355. [Google Scholar] [CrossRef]
- Radford, A.D.; Chapman, D.; Dixon, L.; Chantrey, J.; Darby, A.C.; Hall, N. Application of next-generation sequencing technologies in virology. J. Gen. Virol. 2012, 93, 1853–1868. [Google Scholar] [CrossRef]
- Chiu, C.Y. Viral pathogen discovery. Curr. Opin Microbiol. 2013, 16, 468–478. [Google Scholar] [CrossRef]
- Kapuscinski, M.L.; Bergren, N.A.; Russell, B.J.; Lee, J.S.; Borland, E.M.; Hartman, D.A.; King, D.C.; Hughes, H.R.; Burkhalter, K.L.; Kading, R.C.; et al. Genomic characterization of 99 viruses from the bunyavirus families Nairoviridae, Peribunyaviridae, and Phenuiviridae, including 35 previously unsequenced viruses. PLoS Pathog. 2021, 17, e1009315. [Google Scholar] [CrossRef]
- Greninger, A.L. A decade of RNA virus metagenomics is (not) enough. Virus Res. 2018, 244, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.J.; Chen, X.; Li, C.X.; Qin, X.C.; Li, J.; Cao, J.P.; Eden, J.S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Wolf, Y.I.; Silas, S.; Wang, Y.; Wu, S.; Bocek, M.; Kazlauskas, D.; Krupovic, M.; Fire, A.; Dolja, V.V.; Koonin, E.V. Doubling of the known set of RNA viruses by metagenomic analysis of an aquatic virome. Nat. Microbiol. 2020, 5, 1262–1270. [Google Scholar] [CrossRef]
- Rodgers, M.A.; Vallari, A.S.; Yamaguchi, J.; Holzmayer, V.; Harris, B.; Toure-Kane, C.; Mboup, S.; Badreddine, S.; Mcarthur, C.; Ndembi, N.; et al. ARCHITECT HIV Combo Ag/Ab and RealTime HIV-1 Assays Detect Diverse HIV Strains in Clinical Specimens. AIDS Res. Hum. Retrovir. 2018, 34, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Achari, A.; Federman, S.; Yu, G.; Somasekar, S.; Bartolo, I.; Yagi, S.; Mbala-Kingebeni, P.; Kapetshi, J.; Ahuka-Mundeke, S.; et al. Metagenomic sequencing with spiked primer enrichment for viral diagnostics and genomic surveillance. Nat. Microbiol. 2020, 5, 443–454. [Google Scholar] [CrossRef]
- Butler, E.K.; Rodgers, M.A.; Coller, K.E.; Barnaby, D.; Krilich, E.; Olivo, A.; Cassidy, M.; Mbanya, D.; Kaptue, L.; Ndembi, N.; et al. High prevalence of hepatitis delta virus in Cameroon. Sci. Rep. 2018, 8, 11617. [Google Scholar] [CrossRef]
- Pour, M.; James, L.; Singh, K.; Mampunza, S.; Baer, F.; Scott, J.; Berg, M.G.; Rodgers, M.A.; Cloherty, G.A.; Hackett, J., Jr.; et al. Increased HIV in Greater Kinshasa Urban Health Zones: Democratic Republic of Congo (2017–2018). AIDS Res. Ther. 2020, 17, 67. [Google Scholar] [CrossRef]
- Berg, M.G.; Olivo, A.; Harris, B.J.; Rodgers, M.A.; James, L.; Mampunza, S.; Niles, J.; Baer, F.; Yamaguchi, J.; Kaptue, L.; et al. A high prevalence of potential HIV elite controllers identified over 30 years in Democratic Republic of Congo. EBioMedicine 2021, 65, 103258. [Google Scholar] [CrossRef]
- Coller, K.E.; Butler, E.K.; Luk, K.C.; Rodgers, M.A.; Cassidy, M.; Gersch, J.; McNamara, A.L.; Kuhns, M.C.; Dawson, G.J.; Kaptue, L.; et al. Development and performance of prototype serologic and molecular tests for hepatitis delta infection. Sci. Rep. 2018, 8, 2095. [Google Scholar] [CrossRef]
- Naccache, S.N.; Federman, S.; Veeraraghavan, N.; Zaharia, M.; Lee, D.; Samayoa, E.; Bouquet, J.; Greninger, A.L.; Luk, K.C.; Enge, B.; et al. A cloud-compatible bioinformatics pipeline for ultrarapid pathogen identification from next-generation sequencing of clinical samples. Genome Res. 2014, 24, 1180–1192. [Google Scholar] [CrossRef]
- Zaharia, M.; Bolosky, W.J.; Curtis, K.; Fox, A.; Patterson, D.; Shenker, S.; Stoica, I.; Karp, R.M.; Sittler, T. Faster and More Accurate Sequence Alignment with SNAP. arXiv 2011, arXiv:1111.5572. [Google Scholar]
- Zhao, Y.; Tang, H.; Ye, Y. RAPSearch2: A fast and memory-efficient protein similarity search tool for next-generation sequencing data. Bioinformatics 2012, 28, 125–126. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.T.; Wong, K.; Jackman, S.D.; Schein, J.E.; Jones, S.J.M.; Birol, İ. ABySS: A parallel assembler for short read sequence data. Genome Res. 2009, 19, 1117–1123. [Google Scholar] [CrossRef] [PubMed]
- Treangen, T.J.; Sommer, D.D.; Angly, F.E.; Koren, S.; Pop, M. Next Generation Sequence Assembly with AMOS. Curr. Protoc. Bioinform. 2011, 33, 11.8.1–11.8.18. [Google Scholar] [CrossRef]
- Zhang, Z.; Schwartz, S.; Wagner, L.; Miller, W. A Greedy Algorithm for Aligning DNA Sequences. J. Comput. Biol. 2000, 7, 203–214. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Takahashi, K.; Nei, M. Efficiencies of fast algorithms of phylogenetic inference under the criteria of maximum parsimony, minimum evolution, and maximum likelihood when a large number of sequences are used. Mol. Biol. Evol. 2000, 17, 1251–1258. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Richter, D.C.; Rausch, C.; Dezulian, T.; Franz, M.; Rupp, R. Dendroscope: An interactive viewer for large phylogenetic trees. BMC Bioinform. 2007, 8, 460. [Google Scholar] [CrossRef]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.Y. GGTREE: An r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Kapoor, A.; Simmonds, P.; Lipkin, W.I.; Zaidi, S.; Delwart, E. Use of Nucleotide Composition Analysis To Infer Hosts for Three Novel Picorna-Like Viruses. J. Virol. 2010, 84, 10322–10328. [Google Scholar] [CrossRef] [PubMed]
- Oude Munnink, B.B.; Phan, M.V.T.; Consortium, V.; Simmonds, P.; Koopmans, M.P.G.; Kellam, P.; van der Hoek, L.; Cotten, M. Characterization of Posa and Posa-like virus genomes in fecal samples from humans, pigs, rats, and bats collected from a single location in Vietnam. Virus Evol. 2017, 3, vex022. [Google Scholar] [CrossRef] [PubMed]
- Mollentze, N.; Babayan, S.A.; Streicker, D.G. Identifying and prioritizing potential human-infecting viruses from their genome sequences. PLoS Biol. 2021, 19, e3001390. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Bezerra, R.S.; Bitencourt, H.T.; Covas, D.T.; Kashima, S.; Slavov, S.N. Metagenomic identification of human Gemykibivirus-2 (HuGkV-2) in parenterally infected blood donors from the Brazilian Amazon. Int. J. Infect. Dis. 2020, 98, 249–251. [Google Scholar] [CrossRef]
- Siqueira, J.; Curty, G.; Xutao, D.; Hofer, C.; Machado, E.; Seuánez, H.; Soares, M.; Delwart, E.; Soares, E. Composite Analysis of the Virome and Bacteriome of HIV/HPV Co-Infected Women Reveals Proxies for Immunodeficiency. Viruses 2019, 11, 422. [Google Scholar] [CrossRef]
- Phan, T.G.; Mori, D.; Deng, X.; Rajindrajith, S.; Ranawaka, U.; Fan Ng, T.F.; Bucardo-Rivera, F.; Orlandi, P.; Ahmed, K.; Delwart, E. Small circular single stranded DNA viral genomes in unexplained cases of human encephalitis, diarrhea, and in untreated sewage. Virology 2015, 482, 98–104. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.D.; Vasilakis, N.; Tian, J.H.; Li, C.X.; Chen, L.J.; Eastwood, G.; Diao, X.N.; Chen, M.H.; Chen, X.; et al. Divergent Viruses Discovered in Arthropods and Vertebrates Revise the Evolutionary History of the Flaviviridae and Related Viruses. J. Virol. 2016, 90, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Jia, N.; Liu, H.B.; Ni, X.B.; Bell-Sakyi, L.; Zheng, Y.C.; Song, J.L.; Li, J.; Jiang, B.G.; Wang, Q.; Sun, Y.; et al. Emergence of human infection with Jingmen tick virus in China: A retrospective study. EBioMedicine 2019, 43, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.H.; Che, X.; Garcia, J.A.; Klena, J.D.; Lee, B.; Muller, D.; Ulrich, W.; Corrigan, R.M.; Nichol, S.; Jain, K.; et al. Viral Diversity of House Mice in New York City. mBio 2018, 9, e01354-17. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.L.; Moore, J.S. Nodaviruses of Invertebrates and Fish (Nodaviridae). In Encyclopedia of Virology; Bamford, D.H., Zuckerman, M., Eds.; Academic Press: Oxford, UK, 2021; pp. 819–826. [Google Scholar]
- Cordey, S.; Laubscher, F.; Hartley, M.-A.; Junier, T.; Pérez-Rodriguez, F.J.; Keitel, K.; Vieille, G.; Samaka, J.; Mlaganile, T.; Kagoro, F.; et al. Detection of dicistroviruses RNA in blood of febrile Tanzanian children. Emerg. Microbes Infect. 2019, 8, 613–623. [Google Scholar] [CrossRef]
- Phan, T.G.; Del Valle Mendoza, J.; Sadeghi, M.; Altan, E.; Deng, X.; Delwart, E. Sera of Peruvians with fever of unknown origins include viral nucleic acids from non-vertebrate hosts. Virus Genes 2018, 54, 33–40. [Google Scholar] [CrossRef]
- Zhao, L.; Rosario, K.; Breitbart, M.; Duffy, S. Chapter Three-Eukaryotic Circular Rep-Encoding Single-Stranded DNA (CRESS DNA) Viruses: Ubiquitous Viruses With Small Genomes and a Diverse Host Range. In Advances in Virus Research; Kielian, M., Mettenleiter, T.C., Roossinck, M.J., Eds.; Academic Press: Cambridge, MA, USA, 2019; Volume 103, pp. 71–133. [Google Scholar]
- Gainor, K.; Becker, A.A.M.J.; Malik, Y.S.; Ghosh, S. Detection and Complete Genome Analysis of Circoviruses and Cycloviruses in the Small Indian Mongoose (Urva auropunctata): Identification of Novel Species. Viruses 2021, 13, 1700. [Google Scholar] [CrossRef]
- le Tan, V.; van Doorn, H.R.; Nghia, H.D.; Chau, T.T.; le Tu, T.P.; de Vries, M.; Canuti, M.; Deijs, M.; Jebbink, M.F.; Baker, S.; et al. Identification of a new cyclovirus in cerebrospinal fluid of patients with acute central nervous system infections. mBio 2013, 4, e00231-13. [Google Scholar]
- Oude Munnink, B.B.; Cotten, M.; Canuti, M.; Deijs, M.; Jebbink, M.F.; van Hemert, F.J.; Phan, M.V.; Bakker, M.; Jazaeri Farsani, S.M.; Kellam, P.; et al. A Novel Astrovirus-Like RNA Virus Detected in Human Stool. Virus Evol. 2016, 2, vew005. [Google Scholar] [CrossRef]
- Paysan-Lafosse, T.; Blum, M.; Chuguransky, S.; Grego, T.; Pinto, B.L.; Salazar, G.A.; Bileschi, M.L.; Bork, P.; Bridge, A.; Colwell, L.; et al. InterPro in 2022. Nucleic Acids Res. 2023, 51, D418–D427. [Google Scholar] [CrossRef]
- Karlin, S.; Mrázek, J. Compositional differences within and between eukaryotic genomes. Proc. Natl. Acad. Sci. USA 1997, 94, 10227–10232. [Google Scholar] [CrossRef]
- Lipkin, W.I. The changing face of pathogen discovery and surveillance. Nat. Rev. Microbiol. 2013, 11, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Taylor, J.; Lin, V.; Altman, T.; Barbera, P.; Meleshko, D.; Lohr, D.; Novakovsky, G.; Buchfink, B.; Al-Shayeb, B.; et al. Petabase-scale sequence alignment catalyses viral discovery. Nature 2022, 602, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Greninger, A.L. The challenge of diagnostic metagenomics. Expert Rev. Mol. Diagn. 2018, 18, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Chiu, C. The Role of Metagenomics and Next-Generation Sequencing in Infectious Disease Diagnosis. Clin. Chem. 2021, 68, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.Y.; Miller, S.A. Clinical metagenomics. Nat. Rev. Genet. 2019, 20, 341–355. [Google Scholar] [CrossRef]
- Sanjuán, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral Mutation Rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef]
- Yinda, C.K.; Ghogomu, S.M.; Conceicao-Neto, N.; Beller, L.; Deboutte, W.; Vanhulle, E.; Maes, P.; Van Ranst, M.; Matthijnssens, J. Cameroonian fruit bats harbor divergent viruses, including rotavirus H, bastroviruses, and picobirnaviruses using an alternative genetic code. Virus Evol. 2018, 4, vey008. [Google Scholar] [CrossRef]
- Li, L.; Victoria, J.G.; Wang, C.; Jones, M.; Fellers, G.M.; Kunz, T.H.; Delwart, E. Bat Guano Virome: Predominance of Dietary Viruses from Insects and Plants plus Novel Mammalian Viruses. J. Virol. 2010, 84, 6955–6965. [Google Scholar] [CrossRef]
- Wu, Z.; Ren, X.; Yang, L.; Hu, Y.; Yang, J.; He, G.; Zhang, J.; Dong, J.; Sun, L.; Du, J.; et al. Virome Analysis for Identification of Novel Mammalian Viruses in Bat Species from Chinese Provinces. J. Virol. 2012, 86, 10999–11012. [Google Scholar] [CrossRef]
- Ge, X.; Li, Y.; Yang, X.; Zhang, H.; Zhou, P.; Zhang, Y.; Shi, Z. Metagenomic Analysis of Viruses from Bat Fecal Samples Reveals Many Novel Viruses in Insectivorous Bats in China. J. Virol. 2012, 86, 4620–4630. [Google Scholar] [CrossRef]
- Li, Y.; Altan, E.; Reyes, G.; Halstead, B.; Deng, X.; Delwart, E. Virome of Bat Guano from Nine Northern California Roosts. J. Virol. 2021, 95, e01713-20. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.G.; Kapusinszky, B.; Wang, C.; Rose, R.K.; Lipton, H.L.; Delwart, E.L. The Fecal Viral Flora of Wild Rodents. PLoS Pathog. 2011, 7, e1002218. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Lu, L.; Du, J.; Yang, L.; Ren, X.; Liu, B.; Jiang, J.; Yang, J.; Dong, J.; Sun, L.; et al. Comparative analysis of rodent and small mammal viromes to better understand the wildlife origin of emerging infectious diseases. Microbiome 2018, 6, 178. [Google Scholar] [CrossRef] [PubMed]
- Vibin, J.; Chamings, A.; Collier, F.; Klaassen, M.; Nelson, T.M.; Alexandersen, S. Metagenomics detection and characterisation of viruses in faecal samples from Australian wild birds. Sci. Rep. 2018, 8, 8686. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Yang, J.; Lu, S.; Jin, D.; Pu, J.; Wu, S.; Luo, X.L.; Liu, L.; Li, Z.; Xu, J. RNA Virus Diversity in Birds and Small Mammals From Qinghai-Tibet Plateau of China. Front. Microbiol. 2022, 13, 780651. [Google Scholar] [CrossRef]
- Woo, H.J.; Reifman, J. A quantitative quasispecies theory-based model of virus escape mutation under immune selection. Proc. Natl. Acad. Sci. USA 2012, 109, 12980–12985. [Google Scholar] [CrossRef]
- Sadeghi, M.; Altan, E.; Deng, X.; Barker, C.M.; Fang, Y.; Coffey, L.L.; Delwart, E. Virome of >12 thousand Culex mosquitoes from throughout California. Virology 2018, 523, 74–88. [Google Scholar] [CrossRef]
- Dos Anjos, K.; Nagata, T.; Melo, F.L. Complete Genome Sequence of a Novel Bastrovirus Isolated from Raw Sewage. Genome Announc. 2017, 5, e01010-17. [Google Scholar] [CrossRef]
- Nagai, M.; Okabayashi, T.; Akagami, M.; Matsuu, A.; Fujimoto, Y.; Hashem, M.A.; Mekata, H.; Nakao, R.; Matsuno, K.; Katayama, Y.; et al. Metagenomic identification, sequencing, and genome analysis of porcine hepe-astroviruses (bastroviruses) in porcine feces in Japan. Infect. Genet. Evol. 2021, 88, 104664. [Google Scholar] [CrossRef]
- French, R.K.; Filion, A.; Niebuhr, C.N.; Holmes, E.C. Metatranscriptomic Comparison of Viromes in Endemic and Introduced Passerines in New Zealand. Viruses 2022, 14, 1364. [Google Scholar] [CrossRef]
- Bonny, P.; Schaeffer, J.; Besnard, A.; Desdouits, M.; Ngang, J.J.E.; Le Guyader, F.S. Human and Animal RNA Virus Diversity Detected by Metagenomics in Cameroonian Clams. Front. Microbiol. 2021, 12, 770385. [Google Scholar] [CrossRef]
- Richard, J.C.; Leis, E.M.; Dunn, C.D.; Harris, C.; Agbalog, R.E.; Campbell, L.J.; Knowles, S.; Waller, D.L.; Putnam, J.G.; Goldberg, T.L. Freshwater Mussels Show Elevated Viral Richness and Intensity during a Mortality Event. Viruses 2022, 14, 2603. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Kapoor, A.; Slikas, B.; Bamidele, O.S.; Wang, C.; Shaukat, S.; Masroor, M.A.; Wilson, M.L.; Ndjango, J.-B.N.; Peeters, M.; et al. Multiple Diverse Circoviruses Infect Farm Animals and Are Commonly Found in Human and Chimpanzee Feces. J. Virol. 2010, 84, 1674–1682. [Google Scholar] [CrossRef] [PubMed]
- Thi Kha Tu, N.; Thi Thu Hong, N.; Thi Han Ny, N.; My Phuc, T.; Thi Thanh Tam, P.; Doorn, H.R.V.; Dang Trung Nghia, H.; Thao Huong, D.; An Han, D.; Thi Thu Ha, L.; et al. The Virome of Acute Respiratory Diseases in Individuals at Risk of Zoonotic Infections. Viruses 2020, 12, 960. [Google Scholar] [CrossRef] [PubMed]
- Dayaram, A.; Potter, K.A.; Moline, A.B.; Rosenstein, D.D.; Marinov, M.; Thomas, J.E.; Breitbart, M.; Rosario, K.; Argüello-Astorga, G.R.; Varsani, A. High global diversity of cycloviruses amongst dragonflies. J. Gen. Virol. 2013, 94, 1827–1840. [Google Scholar] [CrossRef]
- Li, L.; Shan, T.; Soji, O.B.; Alam, M.M.; Kunz, T.H.; Zaidi, S.Z.; Delwart, E. Possible cross-species transmission of circoviruses and cycloviruses among farm animals. J. Gen. Virol. 2011, 92, 768–772. [Google Scholar] [CrossRef]
- Sauvage, V.; Gomez, J.; Barray, A.; Vandenbogaert, M.; Boizeau, L.; Tagny, C.T.; Rakoto, O.; Bizimana, P.; Guitteye, H.; Cire, B.B.; et al. High prevalence of cyclovirus Vietnam (CyCV-VN) in plasma samples from Madagascan healthy blood donors. Infect. Genet. Evol. 2018, 66, 9–12. [Google Scholar] [CrossRef]
- Phan, T.G.; Luchsinger, V.; Avendaño, L.F.; Deng, X.; Delwart, E. Cyclovirus in nasopharyngeal aspirates of Chilean children with respiratory infections. J. Gen. Virol. 2014, 95, 922–927. [Google Scholar] [CrossRef]
- Smits, S.L.; Zijlstra, E.E.; Van Hellemond, J.J.; Schapendonk, C.M.E.; Bodewes, R.; Schürch, A.C.; Haagmans, B.L.; Osterhaus, A.D.M.E. Novel Cyclovirus in Human Cerebrospinal Fluid, Malawi, 2010–2011. Emerg. Infect. Dis. 2013, 19, 1511–1513. [Google Scholar] [CrossRef]
- Capozza, P.; Lanave, G.; Diakoudi, G.; Pellegrini, F.; Cardone, R.; Vasinioti, V.I.; Decaro, N.; Elia, G.; Catella, C.; Alberti, A.; et al. Diversity of CRESS DNA Viruses in Squamates Recapitulates Hosts Dietary and Environmental Sources of Exposure. Microbiol. Spectr. 2022, 10, e0078022. [Google Scholar] [CrossRef]
- Averhoff, F.; Berg, M.; Rodgers, M.; Osmanov, S.; Luo, X.; Anderson, M.; Meyer, T.; Landay, A.; Gamkrelidze, A.; Kallas, E.G.; et al. The Abbott Pandemic Defense Coalition: A unique multisector approach adds to global pandemic preparedness efforts. Int. J. Infect. Dis. 2022, 117, 356–360. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orf, G.S.; Olivo, A.; Harris, B.; Weiss, S.L.; Achari, A.; Yu, G.; Federman, S.; Mbanya, D.; James, L.; Mampunza, S.; et al. Metagenomic Detection of Divergent Insect- and Bat-Associated Viruses in Plasma from Two African Individuals Enrolled in Blood-Borne Surveillance. Viruses 2023, 15, 1022. https://doi.org/10.3390/v15041022
Orf GS, Olivo A, Harris B, Weiss SL, Achari A, Yu G, Federman S, Mbanya D, James L, Mampunza S, et al. Metagenomic Detection of Divergent Insect- and Bat-Associated Viruses in Plasma from Two African Individuals Enrolled in Blood-Borne Surveillance. Viruses. 2023; 15(4):1022. https://doi.org/10.3390/v15041022
Chicago/Turabian StyleOrf, Gregory S., Ana Olivo, Barbara Harris, Sonja L. Weiss, Asmeeta Achari, Guixia Yu, Scot Federman, Dora Mbanya, Linda James, Samuel Mampunza, and et al. 2023. "Metagenomic Detection of Divergent Insect- and Bat-Associated Viruses in Plasma from Two African Individuals Enrolled in Blood-Borne Surveillance" Viruses 15, no. 4: 1022. https://doi.org/10.3390/v15041022
APA StyleOrf, G. S., Olivo, A., Harris, B., Weiss, S. L., Achari, A., Yu, G., Federman, S., Mbanya, D., James, L., Mampunza, S., Chiu, C. Y., Rodgers, M. A., Cloherty, G. A., & Berg, M. G. (2023). Metagenomic Detection of Divergent Insect- and Bat-Associated Viruses in Plasma from Two African Individuals Enrolled in Blood-Borne Surveillance. Viruses, 15(4), 1022. https://doi.org/10.3390/v15041022