
First Discovery of Phenuiviruses within Diverse RNA Viromes of Asiatic Toad (Bufo gargarizans) by Metagenomics Sequencing

 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. RNA Extraction
2.3. RNA Library Construction and Sequencing
2.4. Virus Discovery
2.5. Genomic and Phylogenetic Analysis
2.6. Viral Prevalence Calculation
2.7. Virus Isolation
3. Results
3.1. Sampling Results
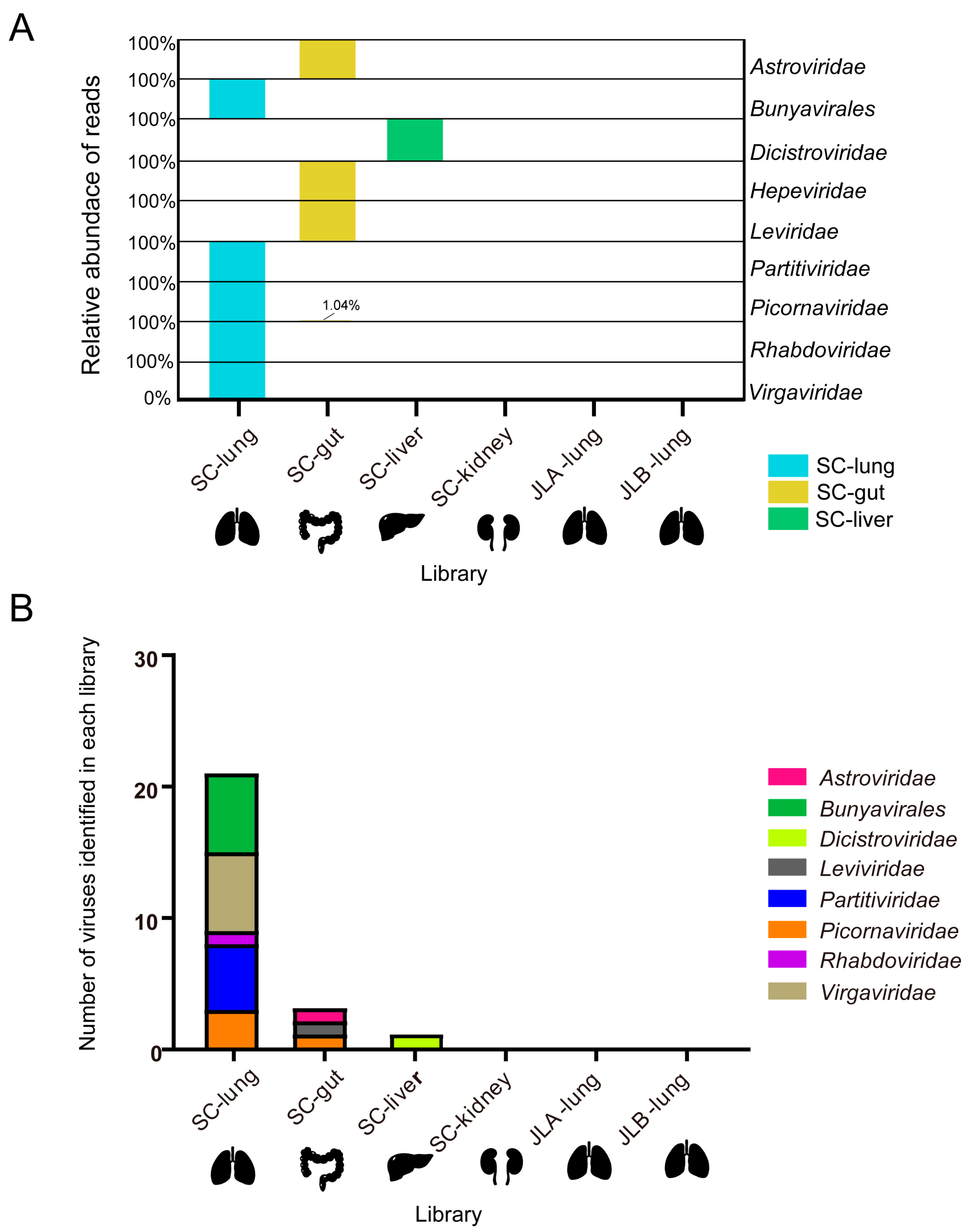
3.2. Overview of RNA Viromes in Asiatic Toad
3.3. Characterization of Novel RNA Viruses in the Asiatic Toad
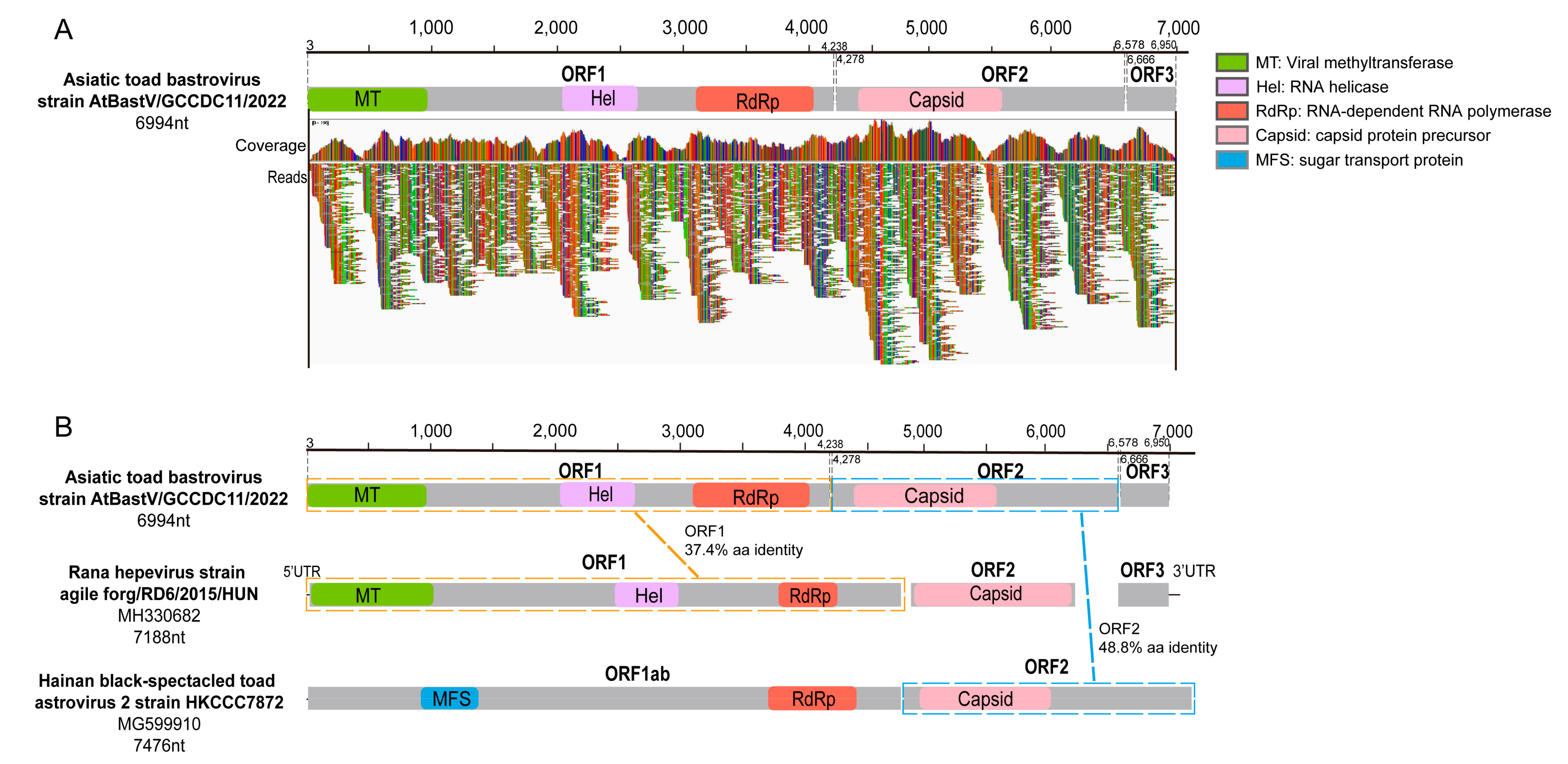
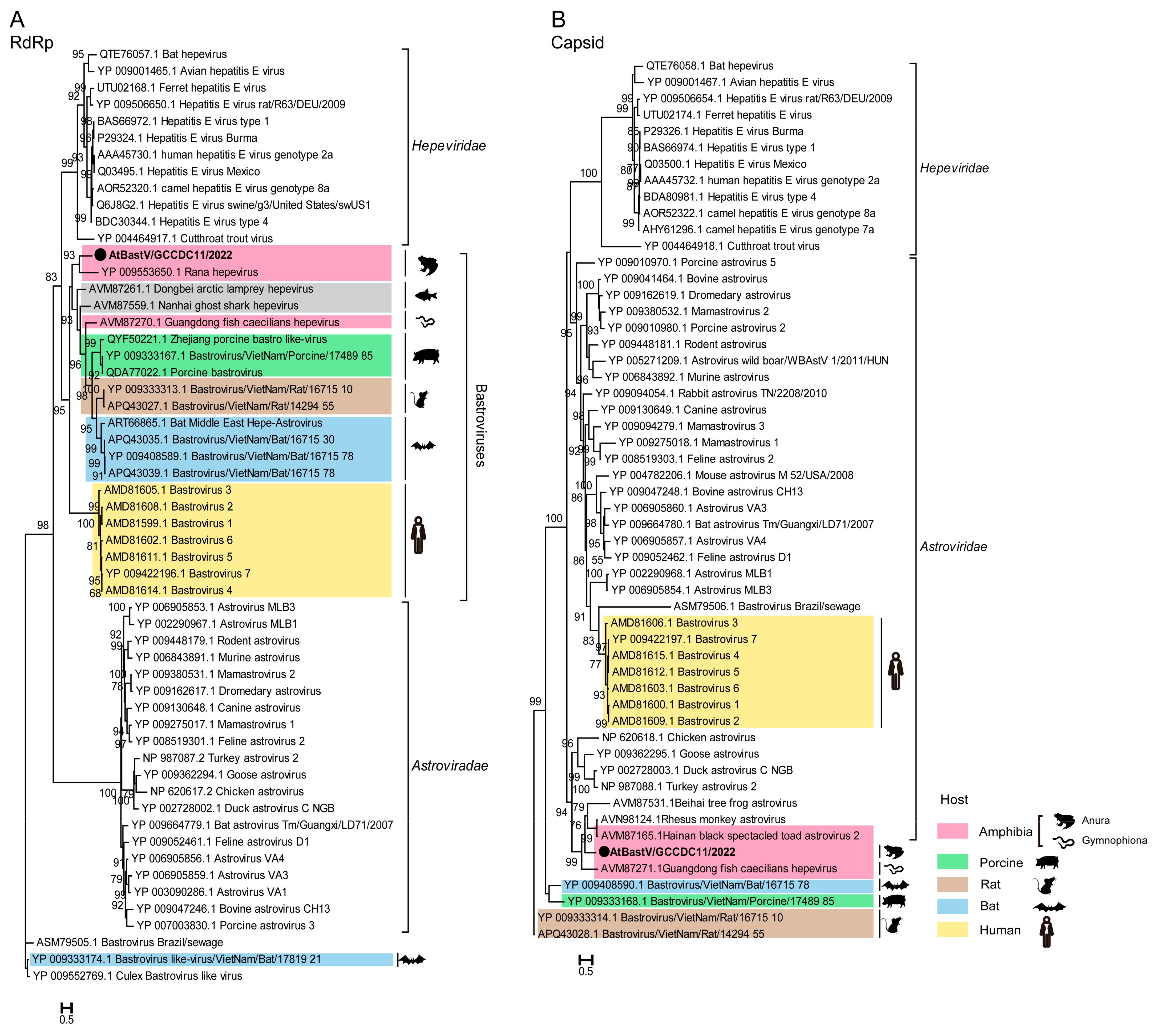
3.3.1. Bastrovirus
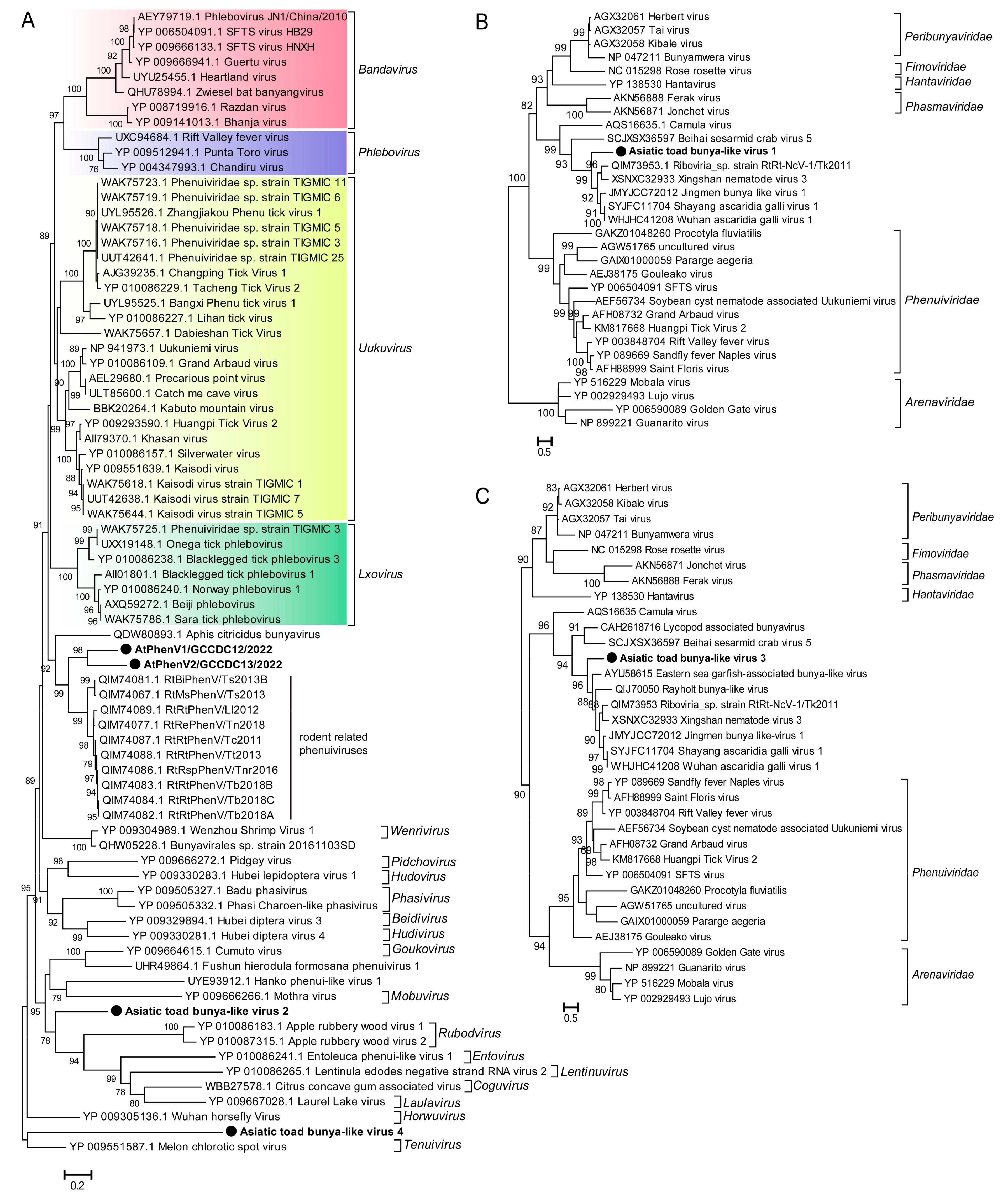
3.3.2. Bunyaviruses
3.3.3. Rhabdovirus
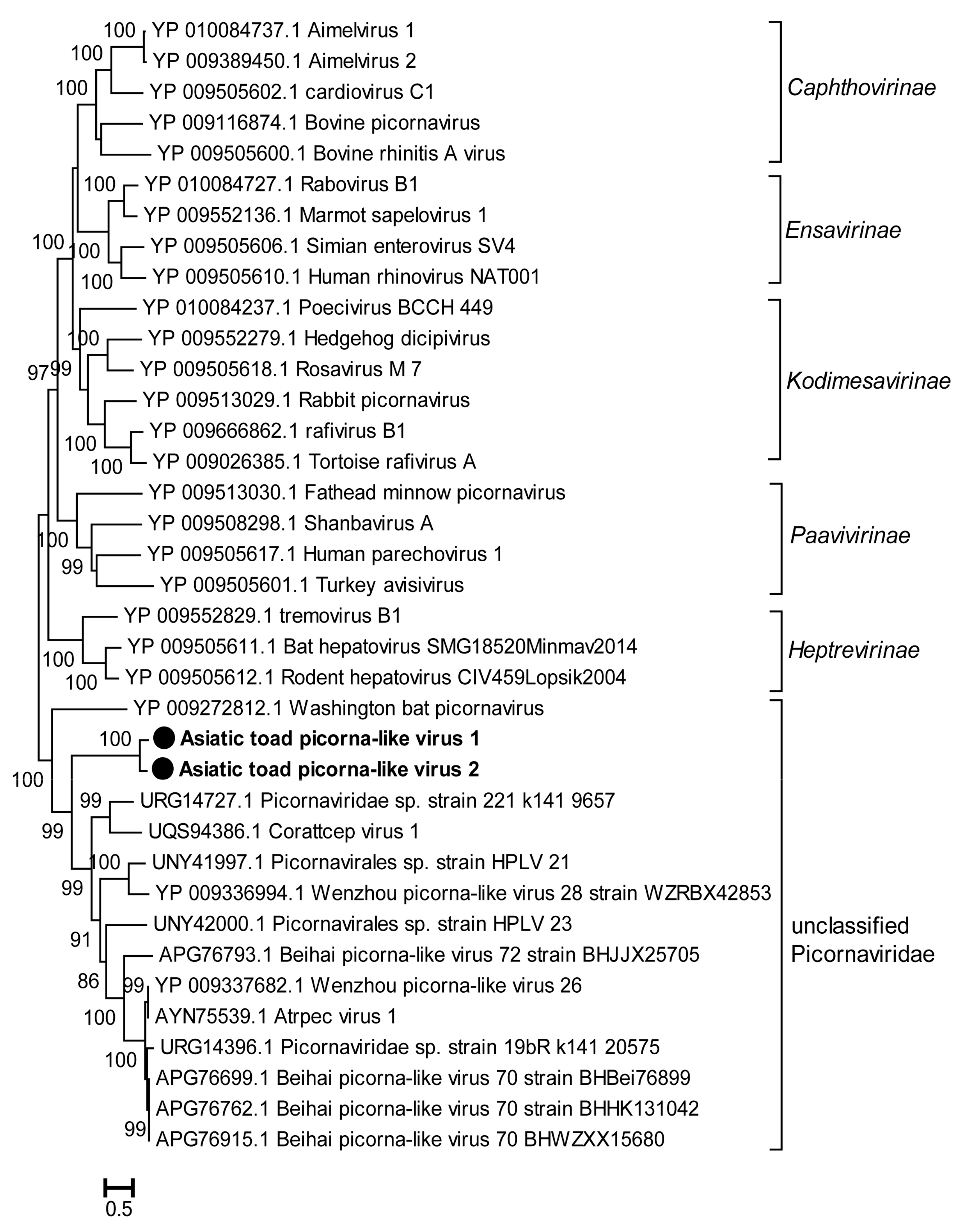
3.3.4. Picornaviruses and Other Invertebrate RNA Viruses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, W.J.; Wu, G. Convincing the confidence to conquer COVID-19: From epidemiological intervention to laboratory investigation. Biosaf. Health 2020, 2, 185–186. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.J.; Wu, Y.; Bi, Y.; Shi, W.; Wang, D.; Shi, Y.; Gao, G.F. Emerging HxNy influenza A viruses. Cold Spring Harb. Perspect. Med. 2022, 12, a038406. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.E.; Patel, N.G.; Levy, M.A.; Storeygard, A.; Balk, D.; Gittleman, J.L.; Daszak, P. Global trends in emerging infectious diseases. Nature 2008, 451, 990–993. [Google Scholar] [CrossRef] [PubMed]
- Morens, D.M.; Folkers, G.K.; Fauci, A.S. The challenge of emerging and re-emerging infectious diseases. Nature 2004, 430, 242–249. [Google Scholar] [CrossRef]
- Dobson Andrew, P.; Pimm Stuart, L.; Hannah, L.; Kaufman, L.; Ahumada Jorge, A.; Ando Amy, W.; Bernstein, A.; Busch, J.; Daszak, P.; Engelmann, J.; et al. Ecology and economics for pandemic prevention. Science 2020, 369, 379–381. [Google Scholar] [CrossRef]
- Wang, X.; Wu, F.; Zhao, X.; Zhang, X.; Wang, J.; Niu, L.; Liang, W.; Leung, K.M.Y.; Giesy, J.P. Enlightenment from the COVID-19 Pandemic: The Roles of Environmental Factors in Future Public Health Emergency Response. Engineering 2021, 8, 108–115. [Google Scholar] [CrossRef]
- Edwards, R.A.; Rohwer, F. Viral metagenomics. Nat. Rev. Microbiol. 2005, 3, 504–510. [Google Scholar] [CrossRef]
- Finkbeiner, S.R.; Allred, A.F.; Tarr, P.I.; Klein, E.J.; Kirkwood, C.D.; Wang, D. Metagenomic analysis of human diarrhea: Viral detection and discovery. PLoS Pathog. 2008, 4, e1000011. [Google Scholar] [CrossRef]
- Adams, I.P.; Glover, R.H.; Monger, W.A.; Mumford, R.; Jackeviciene, E.; Navalinskiene, M.; Samuitiene, M.; Boonham, N. Next-generation sequencing and metagenomic analysis: A universal diagnostic tool in plant virology. Mol. Plant Pathol. 2009, 10, 537–545. [Google Scholar] [CrossRef]
- Tang, P.; Chiu, C. Metagenomics for the discovery of novel human viruses. Future Microbiol. 2010, 5, 177–189. [Google Scholar] [CrossRef]
- Kieft, K.; Anantharaman, K. Virus genomics: What is being overlooked? Curr. Opin. Virol. 2022, 53, 101200. [Google Scholar] [CrossRef]
- Zhang, Y.Z.; Wu, W.C.; Shi, M.; Holmes, E.C. The diversity, evolution and origins of vertebrate RNA viruses. Curr. Opin. Virol. 2018, 31, 9–16. [Google Scholar] [CrossRef]
- Parry, R.; Wille, M.; Turnbull, O.M.H.; Geoghegan, J.L.; Holmes, E.C. Divergent influenza-like viruses of amphibians and fish support an ancient evolutionary sssociation. Viruses 2020, 12, 1042. [Google Scholar] [CrossRef]
- Essbauer, S.; Ahne, W. Viruses of lower vertebrates. J. Vet. Med. B Infect. Dis. Vet. Public Health 2001, 48, 403–475. [Google Scholar] [CrossRef]
- Huang, C.; Liu, W.J.; Xu, W.; Jin, T.; Zhao, Y.; Song, J.; Shi, Y.; Ji, W.; Jia, H.; Zhou, Y.; et al. A bat-derived putative cross-family recombinant coronavirus with a reovirus gene. PLoS Pathog. 2016, 12, e1005883. [Google Scholar] [CrossRef]
- Obameso, J.O.; Li, H.; Jia, H.; Han, M.; Zhu, S.; Huang, C.; Zhao, Y.; Zhao, M.; Bai, Y.; Yuan, F.; et al. The persistent prevalence and evolution of cross-family recombinant coronavirus GCCDC1 among a bat population: A two-year follow-up. Sci. China Life Sci. 2017, 60, 1357–1363. [Google Scholar] [CrossRef]
- Li, X.; Wang, L.; Liu, P.; Li, H.; Huo, S.; Zong, K.; Zhu, S.; Guo, Y.; Zhang, L.; Hu, B.; et al. A novel potentially recombinant rodent coronavirus with a polybasic cleavage site in the spike protein. J. Virol. 2021, 95, e0117321. [Google Scholar] [CrossRef]
- Zhou, S.W.; Quan, J.Y.; Li, Z.W.; Ye, G.; Shang, Z.; Chen, Z.P.; Wang, L.; Li, X.Y.; Zhang, X.Q.; Li, J.; et al. Bufadienolides from the eggs of the toad Bufo bufo gargarizans and their antimelanoma activities. J. Nat. Prod. 2021, 84, 1425–1433. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.D.; Chen, X.; Tian, J.H.; Chen, L.J.; Li, K.; Wang, W.; Eden, J.S.; Shen, J.J.; Liu, L.; et al. The evolutionary history of vertebrate RNA viruses. Nature 2018, 556, 197–202. [Google Scholar] [CrossRef]
- Bukhari, K.; Mulley, G.; Gulyaeva, A.A.; Zhao, L.; Shu, G.; Jiang, J.; Neuman, B.W. Description and initial characterization of metatranscriptomic nidovirus-like genomes from the proposed new family Abyssoviridae, and from a sister group to the Coronavirinae, the proposed genus Alphaletovirus. Virology 2018, 524, 160–171. [Google Scholar] [CrossRef]
- Dill, J.A.; Camus, A.C.; Leary, J.H.; Di Giallonardo, F.; Holmes, E.C.; Ng, T.F. Distinct viral lineages from fish and amphibians reveal the complex evolutionary history of Hepadnaviruses. J. Virol. 2016, 90, 7920–7933. [Google Scholar] [CrossRef] [PubMed]
- Debat, H.J.; Ng, T.F.F. Complete genome sequence of a divergent strain of Tibetan frog hepatitis B virus associated with a concave-eared torrent frog (Odorrana tormota). Arch. Virol. 2019, 164, 1727–1732. [Google Scholar] [CrossRef]
- Othman, S.N.; Litvinchuk, S.N.; Maslova, I.; Dahn, H.; Messenger, K.R.; Andersen, D.; Jowers, M.J.; Kojima, Y.; Skorinov, D.V.; Yasumiba, K.; et al. From Gondwana to the Yellow Sea, evolutionary diversifications of true toads Bufo sp. in the Eastern Palearctic and a revisit of species boundaries for Asian lineages. Elife 2022, 11, e70494. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Li, J.; Zheng, Q.; Li, D. A research update on the antitumor effects of active components of Chinese medicine ChanSu. Front. Oncol. 2022, 12, 1014637. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Hong, Y.; Xie, P.; Chen, Y.; Jiang, L.; Yang, Z.; Cao, G.; Chen, Z.; Liu, X.; Chen, Y.; et al. Spatial lipidomics reveals anticancer mechanisms of Bufalin in combination with Cinobufagin in tumor-bearing mice. Front. Pharmacol. 2020, 11, 593815. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Feng, Y.; Chen, X.; Shi, M.; Fu, S.; Yang, W.; Liu, W.J.; Gao, G.F.; Liang, G. Virome of bat-infesting arthropods: Highly divergent viruses in different vectors. J. Virol. 2022, 96, e0146421. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.J.; Chen, X.; Li, C.X.; Qin, X.C.; Li, J.; Cao, J.P.; Eden, J.S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef]
- Wu, Z.; Han, Y.; Liu, B.; Li, H.; Zhu, G.; Latinne, A.; Dong, J.; Sun, L.; Su, H.; Liu, L.; et al. Decoding the RNA viromes in rodent lungs provides new insight into the origin and evolutionary patterns of rodent-borne pathogens in Mainland Southeast Asia. Microbiome 2021, 9, 18. [Google Scholar] [CrossRef]
- Oude Munnink, B.B.; Cotten, M.; Canuti, M.; Deijs, M.; Jebbink, M.F.; van Hemert, F.J.; Phan, M.V.T.; Bakker, M.; Jazaeri Farsani, S.M.; Kellam, P.; et al. A novel astrovirus-like RNA virus detected in human stool. Virus Evol. 2016, 2, vew005. [Google Scholar] [CrossRef]
- Dos Anjos, K.; Nagata, T.; Melo, F.L. Complete genome sequence of a novel bastrovirus isolated from raw sewage. Genome Announc. 2017, 5, e01010-17. [Google Scholar] [CrossRef]
- Bauermann, F.V.; Hause, B.; Buysse, A.R.; Joshi, L.R.; Diel, D.G. Identification and genetic characterization of a porcine hepe-astrovirus (bastrovirus) in the United States. Arch. Virol. 2019, 164, 2321–2326. [Google Scholar] [CrossRef]
- Yinda, C.K.; Ghogomu, S.M.; Conceicao-Neto, N.; Beller, L.; Deboutte, W.; Vanhulle, E.; Maes, P.; Van Ranst, M.; Matthijnssens, J. Cameroonian fruit bats harbor divergent viruses, including rotavirus H, bastroviruses, and picobirnaviruses using an alternative genetic code. Virus Evol. 2018, 4, vey008. [Google Scholar] [CrossRef]
- Bonny, P.; Schaeffer, J.; Besnard, A.; Desdouits, M.; Ngang, J.J.E.; Le Guyader, F.S. Human and animal RNA virus diversity detected by metagenomics in Cameroonian clams. Front. Microbiol. 2021, 12, 770385. [Google Scholar] [CrossRef]
- Reuter, G.; Boros, A.; Toth, Z.; Kapusinszky, B.; Delwart, E.; Pankovics, P. Detection of a novel RNA virus with hepatitis E virus-like non-structural genome organization in amphibian, agile frog (Rana dalmatina) tadpoles. Infect. Genet. Evol. 2018, 65, 112–116. [Google Scholar] [CrossRef]
- Ayhan, N.; Charrel, R.N. An update on Toscana virus distribution, genetics, medical and diagnostic aspects. Clin. Microbiol. Infect. 2020, 26, 1017–1023. [Google Scholar] [CrossRef]
- Linthicum, K.J.; Britch, S.C.; Anyamba, A. Rift Valley Fever: An emerging mosquito-borne disease. Annu. Rev. Entomol. 2016, 61, 395–415. [Google Scholar] [CrossRef]
- Casel, M.A.; Park, S.J.; Choi, Y.K. Severe fever with thrombocytopenia syndrome virus: Emerging novel phlebovirus and their control strategy. Exp. Mol. Med. 2021, 53, 713–722. [Google Scholar] [CrossRef]
- Liu, J.; Sun, Y.; Shi, W.; Tan, S.; Pan, Y.; Cui, S.; Zhang, Q.; Dou, X.; Lv, Y.; Li, X.; et al. The first imported case of Rift Valley fever in China reveals a genetic reassortment of different viral lineages. Emerg. Microbes Infect. 2017, 6, e4. [Google Scholar] [CrossRef]
- Guardado-Calvo, P.; Rey, F.A. The envelope proteins of the Bunyavirales. Adv. Virus Res. 2017, 98, 83–118. [Google Scholar] [CrossRef]
- Walker, P.J.; Firth, C.; Widen, S.G.; Blasdell, K.R.; Guzman, H.; Wood, T.G.; Paradkar, P.N.; Holmes, E.C.; Tesh, R.B.; Vasilakis, N. Evolution of genome size and complexity in the rhabdoviridae. PLoS Pathog. 2015, 11, e1004664. [Google Scholar] [CrossRef]
- Dietzgen, R.G.; Kondo, H.; Goodin, M.M.; Kurath, G.; Vasilakis, N. The family Rhabdoviridae: Mono- and bipartite negative-sense RNA viruses with diverse genome organization and common evolutionary origins. Virus Res. 2017, 227, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.R.; Streicker, D.G.; Schnell, M.J. The spread and evolution of rabies virus: Conquering new frontiers. Nat. Rev. Microbiol. 2018, 16, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Bekal, S.; Domier, L.L.; Niblack, T.L.; Lambert, K.N. Discovery and initial analysis of novel viral genomes in the soybean cyst nematode. J. Gen. Virol. 2011, 92, 1870–1879. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.N.; da Silva, D.C.; Feitosa, L.A.; Furtado, A.P.; Giese, E.G.; de Vasconcelos Melo, F.T. Rhinella marina (Amphibia: Bufonidae) versus Rhabdias paraensis (Nematoda: Rhabdiasidae): Expanding the view on a natural infection. J. Parasitol. 2016, 102, 349–355. [Google Scholar] [CrossRef]
- Zhao, Q.P.; Dong, H.; Han, H.Y.; Wu, Y.L.; Zhu, S.H.; Li, L.J.; Wu, D.; Liang, S.T.; Li, S.; Zhai, X.; et al. A preliminary investigation on the prevalence of parasites in market toads in Shanghai. Chin. J. Animal. Infect. Dis. 2014, 22, 49–53. [Google Scholar]
- Bedard, K.M.; Semler, B.L. Regulation of picornavirus gene expression. Microbes Infect. 2004, 6, 702–713. [Google Scholar] [CrossRef]
- Russo, A.G.; Harding, E.F.; Yan, G.J.H.; Selechnik, D.; Ducatez, S.; DeVore, J.L.; Zhou, J.; Sarma, R.R.; Lee, Y.P.; Richardson, M.F.; et al. Discovery of novel viruses associated with the invasive Cane toad (Rhinella marina) in its native and introduced ranges. Front. Microbiol. 2021, 12, 733631. [Google Scholar] [CrossRef]
- Reuter, G.; Boros, A.; Toth, Z.; Gia Phan, T.; Delwart, E.; Pankovics, P. A highly divergent picornavirus in an amphibian, the smooth newt (Lissotriton vulgaris). J. Gen. Virol. 2015, 96, 2607–2613. [Google Scholar] [CrossRef]
- Pankovics, P.; Boros, A.; Toth, Z.; Phan, T.G.; Delwart, E.; Reuter, G. Genetic characterization of a second novel picornavirus from an amphibian host, smooth newt (Lissotriton vulgaris). Arch. Virol. 2017, 162, 1043–1050. [Google Scholar] [CrossRef]
- Russo, A.G.; Eden, J.S.; Enosi Tuipulotu, D.; Shi, M.; Selechnik, D.; Shine, R.; Rollins, L.A.; Holmes, E.C.; White, P.A. Viral discovery in the invasive Australian Cane toad (Rhinella marina) using metatranscriptomic and genomic approaches. J. Virol. 2018, 92, e00768-18. [Google Scholar] [CrossRef]
- Li, C.X.; Shi, M.; Tian, J.H.; Lin, X.D.; Kang, Y.J.; Chen, L.J.; Qin, X.C.; Xu, J.; Holmes, E.C.; Zhang, Y.Z. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. Elife 2015, 4, e05378. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.D.; Vasilakis, N.; Tian, J.H.; Li, C.X.; Chen, L.J.; Eastwood, G.; Diao, X.N.; Chen, M.H.; Chen, X.; et al. Divergent viruses discovered in arthropods and vertebrates revise the evolutionary history of the Flaviviridae and related viruses. J. Virol. 2016, 90, 659–669. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site | Date of Collection (mm/yyyy) | Individual Number | Tissue (Number) | Library | Raw Reads | Clean Reads | Clean Base Q30 1 | Scaffolds Number | Scaffold Average Length (bp) | N50 2 |
|---|---|---|---|---|---|---|---|---|---|---|
| Sichuan | 4/2022 | 9 | Lung (9) | SC-lung | 172,714,428 | 171,489,864 | 89.7% | 350 | 952.9 | 966 |
| Gut (9) | SC-gut | 133,230,938 | 132,643,352 | 93.3% | 811 | 987.8 | 1009 | |||
| Liver (9) | SC-liver | 158,158,996 | 157,069,570 | 90.2% | 809 | 962.2 | 937 | |||
| Kidney (9) | SC-kidney | 127,191,896 | 126,644,714 | 92.8% | 236 | 974.2 | 1011 | |||
| Jilin | 7/2022 | 8 | Lung (3) | JLA-lung | 94,277,790 | 89,464,230 | 93.1% | 13 | 1290.4 | 1706 |
| Lung (5) | JLB-lung | 96,489,696 | 92,291,648 | 93.3% | 25 | 1582.4 | 1973 |
| Order/Family | Library | Virus Name | Length (bp) | BLASTx Hits on Known Viruses (Blast Amino Acid Identity) | Reads |
|---|---|---|---|---|---|
| Astroviridae | SC-gut | AtBastV/GCCDC11/2022 | 6994 | Rana hepevirus (36.6%) | 301 |
| Bunyavirales | SC-lung | AtPhenV1/GCCDC12/2022 | 2270 | Phenuiviridae sp. strain RtRt-PhenV/Tb2018B (41.6%) | 82 |
| AtPhenV2/GCCDC13/2022 | 2622 | Phenuiviridae sp. strain RtRt-PhenV/Tc2011 (40.1%) | 142 | ||
| Asiatic toad bunya-like virus 1 | 769 | Jingmen bunya-like virus 1 (42.6%) | 27 | ||
| Asiatic toad bunya-like virus 2 | 1752 | Bunyavirales sp. strain 20161103SD (26.0%) | 109 | ||
| Asiatic toad bunya-like virus 3 | 573 | Eastern sea garfish-associated bunya-like virus (40.8%) | 20 | ||
| Asiatic toad bunya-like virus 4 | 781 | Phenuiviridae sp. strain TIGMIC_25 (30.6%) | 36 | ||
| Dicistroviridae | SC-liver | Asiatic toad picorna-like virus 4 | 745 | Dicistroviridae sp. strain XZN128099 (46.1%) | 25 |
| Leviviridae | SC-gut | Asiatic toad levi-like virus | 581 | ssRNA phage AVE015 (51.8%) | 29 |
| Partitiviridae | SC-lung | Wuhan spider virus 10 * | 763 | Wuhan spider virus 10 (95.2%) | 35 |
| Asiatic toad partiti-like virus 1 | 1554 | Xinzhou partiti-like virus 1 (44.8%) | 297 | ||
| Asiatic toad partiti-like virus 2 | 1386 | Xinzhou partiti-like virus 1 (51.7%) | 231 | ||
| Asiatic toad partiti-like virus 3 | 865 | Xinzhou partiti-like virus 1 (48.5%) | 105 | ||
| Picornaviridae | SC-lung | Asiatic toad picorna-like virus 1 | 9550 | Picornaviridae sp. strain 19bR-k141_20575 (34.0%) | 12,849 |
| Asiatic toad picorna-like virus 2 | 3539 | Corattcep virus 1 (26.0%) | 373 | ||
| Asiatic toad picorna-like virus 3 | 1013 | Cripavirus sp. strain s59-k141_1227864 (35.1%) | 94 | ||
| SC-gut | Asiatic toad picorna-like virus 1 | 735 | Wenzhou picorna-like virus 26 (37.3%) | 141 | |
| Rhabdoviridae | SC-lung | Asiatic toad rhabdo-like virus | 5068 | Soybean cyst nematode associated northern cereal mosaic virus (32.1%) | 306 |
| Virgaviridae | SC-lung | Asiatic toad virga-like virus 1 | 2144 | Nelorpivirus dungfly (37.1%) | 167 |
| Asiatic toad virga-like virus 2 | 575 | Clonorsi virus 1 (29.0%) | 46 | ||
| Asiatic toad virga-like virus 3 | 1288 | Blueberry necrotic ring blotch virus (46.0%) | 73 | ||
| Asiatic toad virga-like virus 4 | 930 | Hubei negev-like virus 1 (38.8%) | 60 | ||
| Asiatic toad virga-like virus 5 | 1091 | Bemisia tabaci nege-like virus 1 (31.8%) | 65 | ||
| Asiatic toad virga-like virus 6 | 1077 | Fasciohepa virus 3 (27.2%) | 82 |
| Virus Name | Length (nt) | Protein Size (aa) | BLASTp Hit (Host) | aa Identity |
|---|---|---|---|---|
| AtPhenV1/GCCDC12/2022 | 2270 | 756 | Phenuiviridae sp. strain RtRt-PhenV/Tb2018B (Rattus tanezum) | 41.6% |
| AtPhenV2/GCCDC13/2022 | 2622 | 873 | Phenuiviridae sp. strain RtRt-PhenV/Tc2011 (Rattus tanezum) | 40.4% |
| Asiatic toad bunya-like virus 1 | 769 | 256 | Jingmen bunya-like virus 1 (common pheasant gut roundworm) | 42.6% |
| Asiatic toad bunya-like virus 2 | 1752 | 583 | Kaisodi virus strain TIGMIC_1 (Haemaphysalis montgomeryi) | 28.8% |
| Asiatic toad bunya-like virus 3 | 573 | 190 | Eastern sea garfish-associated bunya-like virus (Hyporhamphus australis) | 40.8% |
| Asiatic toad bunya-like virus 4 | 781 | 259 | Phenuiviridae sp. strain TIGMIC_25 (Dermacentor nuttalli) | 30.6% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Z.; Zhao, H.; Li, Z.; Huang, M.; Si, N.; Zhao, H.; Wei, X.; Sun, B.; Gao, G.F.; Xu, Z.; et al. First Discovery of Phenuiviruses within Diverse RNA Viromes of Asiatic Toad (Bufo gargarizans) by Metagenomics Sequencing. Viruses 2023, 15, 750. https://doi.org/10.3390/v15030750
Chen Z, Zhao H, Li Z, Huang M, Si N, Zhao H, Wei X, Sun B, Gao GF, Xu Z, et al. First Discovery of Phenuiviruses within Diverse RNA Viromes of Asiatic Toad (Bufo gargarizans) by Metagenomics Sequencing. Viruses. 2023; 15(3):750. https://doi.org/10.3390/v15030750
Chicago/Turabian StyleChen, Zhangfu, Haiyu Zhao, Zhongkuan Li, Mengkun Huang, Nan Si, Hui Zhao, Xiaolu Wei, Bo Sun, George F. Gao, Ziqian Xu, and et al. 2023. "First Discovery of Phenuiviruses within Diverse RNA Viromes of Asiatic Toad (Bufo gargarizans) by Metagenomics Sequencing" Viruses 15, no. 3: 750. https://doi.org/10.3390/v15030750
APA StyleChen, Z., Zhao, H., Li, Z., Huang, M., Si, N., Zhao, H., Wei, X., Sun, B., Gao, G. F., Xu, Z., & Liu, W. J. (2023). First Discovery of Phenuiviruses within Diverse RNA Viromes of Asiatic Toad (Bufo gargarizans) by Metagenomics Sequencing. Viruses, 15(3), 750. https://doi.org/10.3390/v15030750