

Evaluating Data Sharing of SARS-CoV-2 Genomes for Molecular Epidemiology across the COVID-19 Pandemic


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
- (a).
- Absolute values of shared SARS-CoV-2 genomes by each individual country and by continent since the beginning of the pandemic.
- (b).
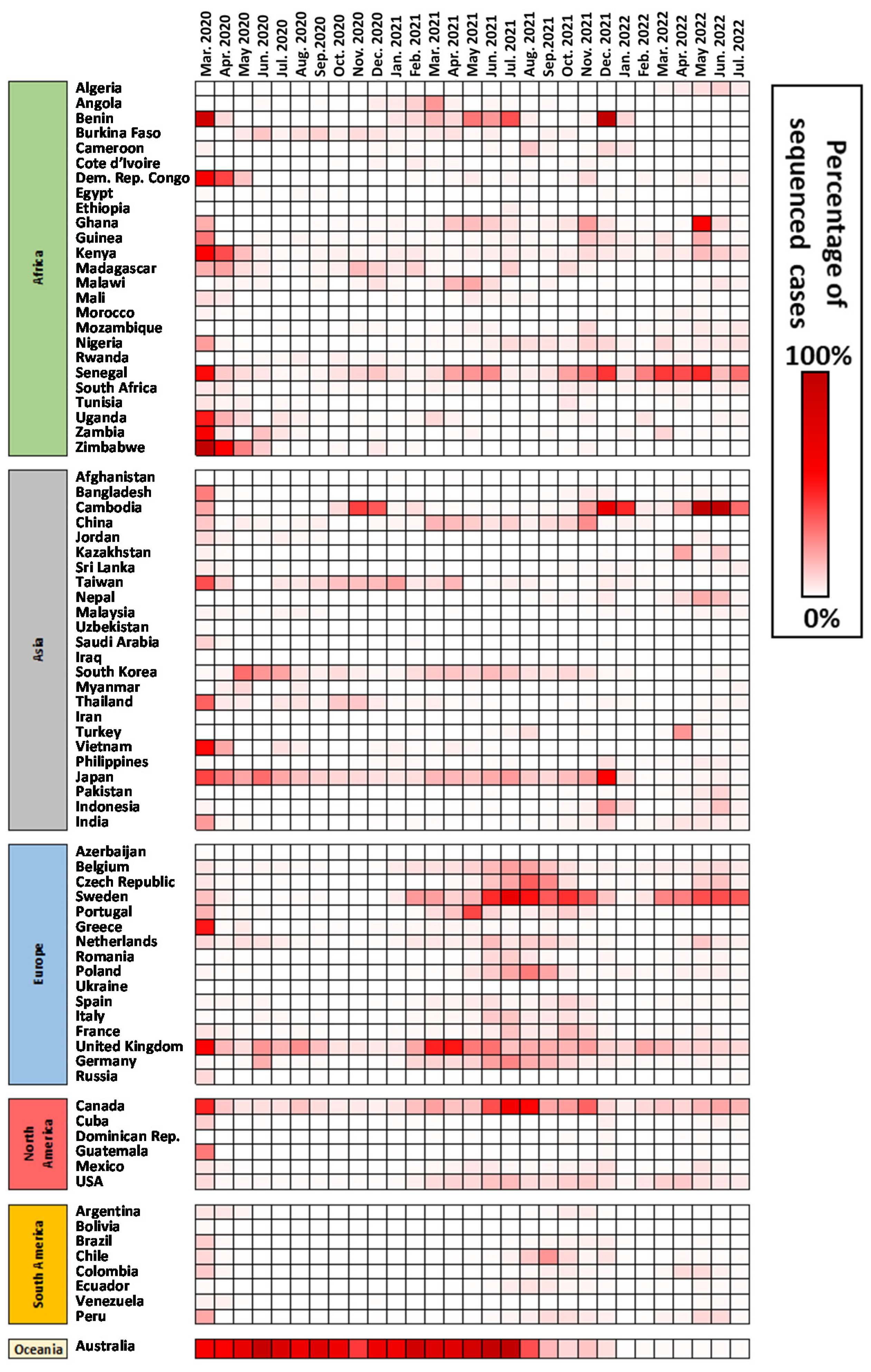
- Total data shared monthly by each individual country and by continent. Data were analyzed from January 2020 until October 2022, although early months and later months in the analysis were not displayed.
- (c).
- Time elapsed between collection date of the SARS-CoV-2 isolate and deposit of the genomic data in the GISAID platform across different continents and countries across different periods of the pandemic (January 2020 until October 2022).
- (d).
- Hypothetical percentage of SARS-CoV-2 genomes were fully characterized given the incidence of COVID-19 at the local level (country) and more general level (continental) across the period in analysis.
- (e).
- A ratio between the percentage of data produced/shared by each country given the number of reported cases in that country against the corresponding percentage of genomes sequenced/shared given the global incidence of COVID-19; this measure provides a view of the relative contribution of each country in data sharing against the average world effort. Again, values are obtained for the full period (January 2020 until October 2022).
- (f).
- Estimated percentage of isolates not sequenced (or hypothetically not shared) of SARS-CoV-2 in each country against the corresponding worldwide number of isolates not characterized during each month of the COVID-19 pandemic (January 2020 until October 2022). While the virus does not seem to display a very high mutation rate [22,23,24], the fact is that each individual case represents a novel evolutionary line where probabilistic novel mutations can emerge. In that context the number of cases, either occurring locally or worldwide, represents the possibility of a novel mutation; from these, a few will be sequenced and characterized at the genomic level while the great majority will not. The value of incidence minus the sequenced genomes provides the number of lineages that could harbor undetected variants each month. The calculated index indicates each country’s worldwide percentage of putative, undetected variants.
- (g).
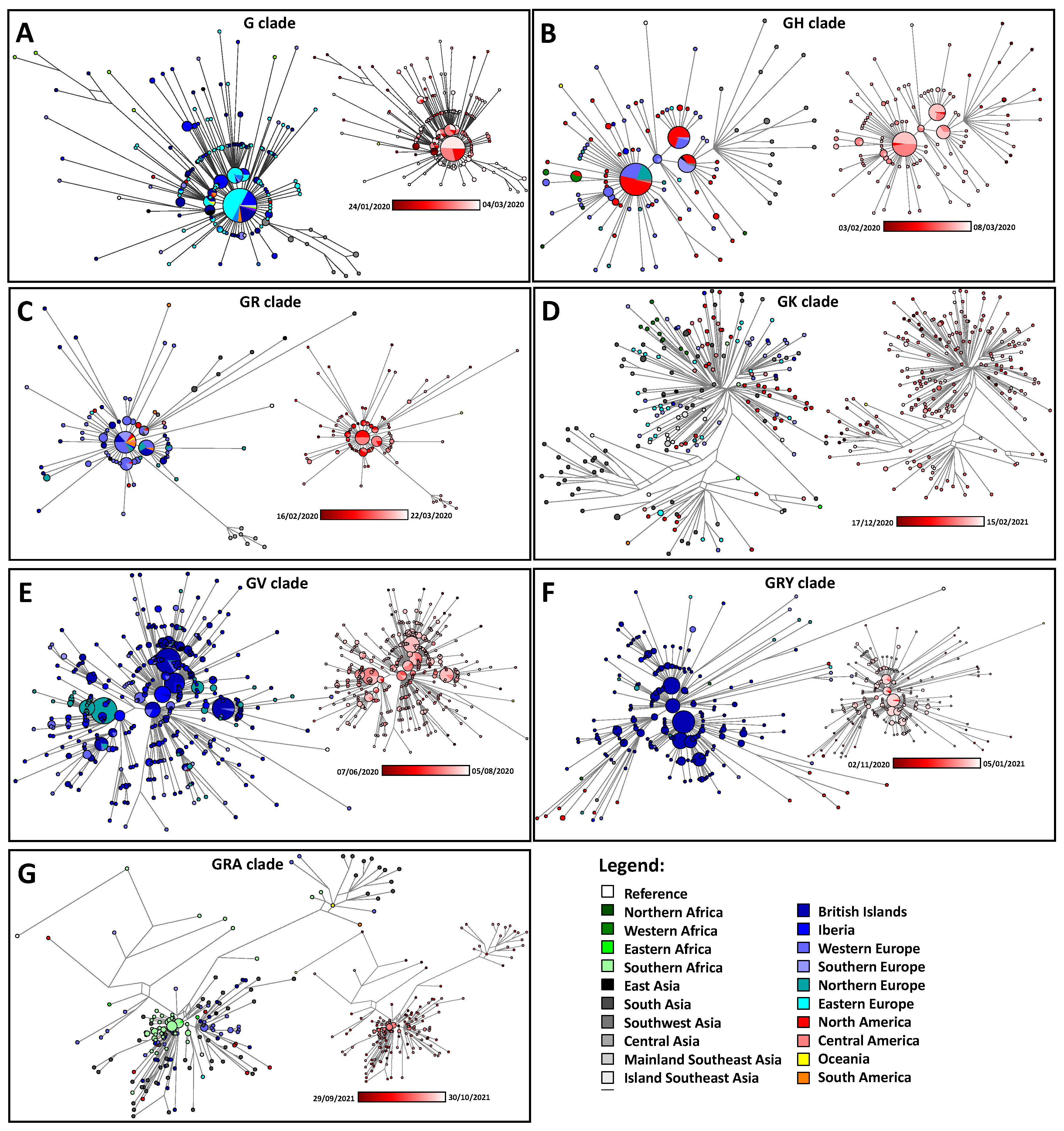
- We collected the early appearance of the major clades, G, GH, GV, GK, GR, GRA and GRY [25], on a global scale. We performed a spatial interpolation using the Kriging algorithm of the Surfer 8 software in order to geographically plot the first appearance of members of each clade according to the data deposited in GISAID. The Kriging algorithm interpolates the best linear unbiased prediction in geostatistics based on the Gaussian process regression. First appearances (for roughly the first 50 days) were plotted geographically. We also checked the phylogenetic positioning of these early cases using the Network software and the reduced median algorithm [26] as we have done before [16]. This also allowed us to correct for mislabeled clades in GISAID. Only genomes of a high coverage were used in the phylogenetic reconstruction. The reduced median algorithm while employed initially in human mitochondrial DNA data [26,27,28] has proved excellent in dealing with molecular epidemiological data [29,30]. The reduced median algorithm ran with the standard threshold of 2.0 with no weighting of the characters.
3. Results
3.1. Overview of Deposited Data in GISAID
3.2. Elapsed Time between Collection of Samples and Data Deposit
3.3. Establishing Risk Scenarios for Undetected Variants
3.4. Exploring the Emergence and Detection of Seven Major Clades throughout the Pandemic
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hunt, R.H.; East, J.E.; Lanas, A.; Malfertheiner, P.; Satsangi, J.; Scarpignato, C.; Webb, G.J. COVID-19 and Gastrointestinal Disease: Implications for the Gastroenterologist. Dig. Dis. 2021, 39, 119–139. [Google Scholar] [CrossRef] [PubMed]
- Pezzini, A.; Padovani, A. Lifting the Mask on Neurological Manifestations of COVID-19. Nat. Rev. Neurol. 2020, 16, 636–644. [Google Scholar] [CrossRef] [PubMed]
- Nasserie, T.; Hittle, M.; Goodman, S.N. Assessment of the Frequency and Variety of Persistent Symptoms among Patients with COVID-19: A Systematic Review. JAMA Netw. Open 2021, 4, e2111417. [Google Scholar] [CrossRef] [PubMed]
- Salamanna, F.; Veronesi, F.; Martini, L.; Landini, M.P.; Fini, M. Post-COVID-19 Syndrome: The Persistent Symptoms at the Post-Viral Stage of the Disease. A Systematic Review of the Current Data. Front. Med. 2021, 8, 653516. [Google Scholar] [CrossRef] [PubMed]
- Barouch, D.H. COVID-19 Vaccines—Immunity, Variants, Boosters. N. Engl. J. Med. 2022, 387, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, E.; Ritchie, H.; Ortiz-Ospina, E.; Roser, M.; Hasell, J.; Appel, C.; Giattino, C.; Rodés-Guirao, L. A Global Database of COVID-19 Vaccinations. Nat. Hum. Behav. 2021, 5, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Liu, Z.; Chen, Z.; Huang, X.; Xu, M.; He, T.; Zhang, Z. The Establishment of Reference Sequence for SARS-CoV-2 and Variation Analysis. J. Med. Virol. 2020, 92, 667–674. [Google Scholar] [CrossRef]
- Banerjee, A.; Doxey, A.C.; Mossman, K.; Irving, A.T. Unraveling the Zoonotic Origin and Transmission of SARS-CoV-2. Trends Ecol. Evol. 2021, 36, 180–184. [Google Scholar] [CrossRef]
- Lau, S.K.P.; Luk, H.K.H.; Wong, A.C.P.; Li, K.S.M.; Zhu, L.; He, Z.; Fung, J.; Chan, T.T.Y.; Fung, K.S.C.; Woo, P.C.Y. Possible Bat Origin of Severe Acute Respiratory Syndrome Coronavirus 2. Emerg. Infect. Dis. 2020, 26, 1542–1547. [Google Scholar] [CrossRef]
- Shu, Y.; McCauley, J. GISAID: Global Initiative on Sharing All Influenza Data—From Vision to Reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef]
- Henschel, A.; Feng, S.F.; Hamoudi, R.A.; Elbait, G.D.; Damiani, E.; Waasia, F.; Tay, G.K.; Mahboub, B.H.; Uddin, M.H.; Acuna, J.; et al. Travel Ban Effects on SARS-CoV-2 Transmission Lineages in the UAE as Inferred by Genomic Epidemiology. PLoS ONE 2022, 17, e0264682. [Google Scholar] [CrossRef] [PubMed]
- Yong, S.E.F.; Anderson, D.E.; Wei, W.E.; Pang, J.; Chia, W.N.; Tan, C.W.; Teoh, Y.L.; Rajendram, P.; Toh, M.P.H.S.; Poh, C.; et al. Connecting Clusters of COVID-19: An Epidemiological and Serological Investigation. Lancet Infect. Dis. 2020, 20, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Didelot, X.; Yang, J.; Wong, G.; Shi, Y.; Liu, W.; Gao, G.F.; Bi, Y. Inference of Person-to-Person Transmission of COVID-19 Reveals Hidden Super-Spreading Events during the Early Outbreak Phase. Nat. Commun. 2020, 11, 5006. [Google Scholar] [CrossRef] [PubMed]
- Brazete, C.; Pinto, M.; Sá, L.; Aguiar, A.; Alves, F.; Duarte, R. Evaluation of the Real-World Effectiveness of Vaccines against COVID-19 at a Local Level: Protocol for a Test-Negative Case–Control Study. Vaccines 2022, 10, 822. [Google Scholar] [CrossRef]
- Gu, H.; Xie, R.; Adam, D.C.; Tsui, J.L.-H.; Chu, D.K.; Chang, L.D.J.; Cheuk, S.S.Y.; Gurung, S.; Krishnan, P.; Ng, D.Y.M.; et al. Genomic Epidemiology of SARS-CoV-2 under an Elimination Strategy in Hong Kong. Nat. Commun. 2022, 13, 736. [Google Scholar] [CrossRef] [PubMed]
- Rito, T.; Richards, M.B.; Pala, M.; Correia-Neves, M.; Soares, P.A. Phylogeography of 27,000 SARS-CoV-2 Genomes: Europe as the Major Source of the COVID-19 Pandemic. Microorganisms 2020, 8, 1678. [Google Scholar] [CrossRef] [PubMed]
- Attwood, S.W.; Hill, S.C.; Aanensen, D.M.; Connor, T.R.; Pybus, O.G. Phylogenetic and Phylodynamic Approaches to Understanding and Combating the Early SARS-CoV-2 Pandemic. Nat. Rev. Genet. 2022, 23, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Quadir, N.; Singh, J.; Alam, A.; Malik, A.A.; Rahman, S.A.; Hira, S.; Ehtesham, N.Z.; Sundar, D.; Hasnain, S.E. Evolution of SARS-CoV-2: BA.4/BA.5 Variants Continues to Pose New Challenges. Viruses 2022, 14, 2610. [Google Scholar] [CrossRef]
- Volz, E.; Mishra, S.; Chand, M.; Barrett, J.C.; Johnson, R.; Geidelberg, L.; Hinsley, W.R.; Laydon, D.J.; Dabrera, G.; O’Toole, Á.; et al. Assessing Transmissibility of SARS-CoV-2 Lineage B.1.1.7 in England. Nature 2021, 593, 266–269. [Google Scholar] [CrossRef]
- Hoffmann, M.; Arora, P.; Groß, R.; Seidel, A.; Hörnich, B.F.; Hahn, A.S.; Krüger, N.; Graichen, L.; Hofmann-Winkler, H.; Kempf, A.; et al. SARS-CoV-2 Variants B.1.351 and P.1 Escape from Neutralizing Antibodies. Cell 2021, 184, 2384–2393.e12. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Lessells, R.J.; Giandhari, J.; Pillay, S.; Msomi, N.; Mlisana, K.; Bhiman, J.N.; von Gottberg, A.; Walaza, S.; et al. Sixteen Novel Lineages of SARS-CoV-2 in South Africa. Nat. Med. 2021, 27, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Callaway, E. The Coronavirus Is Mutating—Does It Matter? Nature 2020, 585, 174–177. [Google Scholar] [CrossRef] [PubMed]
- Vilar, S.; Isom, D.G. One Year of SARS-CoV-2: How Much Has the Virus Changed? Biology 2021, 10, 91. [Google Scholar] [CrossRef] [PubMed]
- De Maio, N.; Walker, C.R.; Turakhia, Y.; Lanfear, R.; Corbett-Detig, R.; Goldman, N. Mutation Rates and Selection on Synonymous Mutations in SARS-CoV-2. Genome Biol. Evol. 2021, 13, evab087. [Google Scholar] [CrossRef] [PubMed]
- Sharif, N.; Alzahrani, K.J.; Ahmed, S.N.; Khan, A.; Banjer, H.J.; Alzahrani, F.M.; Parvez, A.K.; Dey, S.K. Genomic Surveillance, Evolution and Global Transmission of SARS-CoV-2 during 2019–2022. PLoS ONE 2022, 17, e0271074. [Google Scholar] [CrossRef] [PubMed]
- Bandelt, H.J.; Forster, P.; Sykes, B.C.; Richards, M.B. Mitochondrial Portraits of Human Populations Using Median Networks. Genetics 1995, 141, 743–753. [Google Scholar] [CrossRef]
- Gomes, V.; Pala, M.; Salas, A.; Álvarez-Iglesias, V.; Amorim, A.; Gómez-Carballa, A.; Carracedo, Á.; Clarke, D.J.; Hill, C.; Mormina, M.; et al. Mosaic Maternal Ancestry in the Great Lakes Region of East Africa. Hum. Genet. 2015, 134, 1013–1027. [Google Scholar] [CrossRef]
- Richards, M.; Macaulay, V.; Hickey, E.; Vega, E.; Sykes, B.; Guida, V.; Rengo, C.; Sellitto, D.; Cruciani, F.; Kivisild, T.; et al. Tracing European Founder Lineages in the Near Eastern MtDNA Pool. Am. J. Hum. Genet. 2000, 67, 1251–1276. [Google Scholar] [CrossRef]
- Lehmann, T.; Marcet, P.L.; Graham, D.H.; Dahl, E.R.; Dubey, J.P. Globalization and the Population Structure of Toxoplasma Gondii. Proc. Natl. Acad. Sci. USA 2006, 103, 11423–11428. [Google Scholar] [CrossRef]
- Rito, T.; Matos, C.; Carvalho, C.; Machado, H.; Rodrigues, G.; Oliveira, O.; Ferreira, E.; Gonçalves, J.; Maio, L.; Morais, C.; et al. A Complex Scenario of Tuberculosis Transmission Is Revealed through Genetic and Epidemiological Surveys in Porto. BMC Infect. Dis. 2018, 18, 53. [Google Scholar] [CrossRef]
- Inlamea, O.F.; Soares, P.; Ikuta, C.Y.; Heinemann, M.B.; Achá, S.J.; Machado, A.; Ferreira Neto, J.S.; Correia-Neves, M.; Rito, T. Evolutionary Analysis of Mycobacterium Bovis Genotypes across Africa Suggests Co-Evolution with Livestock and Humans. PLoS Negl. Trop. Dis. 2020, 14, e0008081. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Peng, H.; Quinlan, B.D.; Rangarajan, E.S.; Pan, A.; Vanderheiden, A.; Suthar, M.S.; et al. SARS-CoV-2 Spike-Protein D614G Mutation Increases Virion Spike Density and Infectivity. Nat. Commun. 2020, 11, 6013. [Google Scholar] [CrossRef]
- Abdool Karim, S.S.; de Oliveira, T. New SARS-CoV-2 Variants—Clinical, Public Health, and Vaccine Implications. N. Engl. J. Med. 2021, 384, 1866–1868. [Google Scholar] [CrossRef]
- Mohapatra, R.K.; Tiwari, R.; Sarangi, A.K.; Islam, M.R.; Chakraborty, C.; Dhama, K. Omicron (B.1.1.529) Variant of SARS-CoV-2: Concerns, Challenges, and Recent Updates. J. Med. Virol. 2022, 94, 2336–2342. [Google Scholar] [CrossRef]
- Guidance for Representative and Targeted Genomic SARS-CoV-2 Monitoring. Available online: https://www.ecdc.europa.eu/en/publications-data/guidance-representative-and-targeted-genomic-sars-cov-2-monitoring (accessed on 31 December 2022).
- Williams, T.C.; Burgers, W.A. SARS-CoV-2 Evolution and Vaccines: Cause for Concern? Lancet Respir. Med. 2021, 9, 333–335. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rito, T.; Fernandes, P.; Duarte, R.; Soares, P. Evaluating Data Sharing of SARS-CoV-2 Genomes for Molecular Epidemiology across the COVID-19 Pandemic. Viruses 2023, 15, 560. https://doi.org/10.3390/v15020560
Rito T, Fernandes P, Duarte R, Soares P. Evaluating Data Sharing of SARS-CoV-2 Genomes for Molecular Epidemiology across the COVID-19 Pandemic. Viruses. 2023; 15(2):560. https://doi.org/10.3390/v15020560
Chicago/Turabian StyleRito, Teresa, Pedro Fernandes, Raquel Duarte, and Pedro Soares. 2023. "Evaluating Data Sharing of SARS-CoV-2 Genomes for Molecular Epidemiology across the COVID-19 Pandemic" Viruses 15, no. 2: 560. https://doi.org/10.3390/v15020560
APA StyleRito, T., Fernandes, P., Duarte, R., & Soares, P. (2023). Evaluating Data Sharing of SARS-CoV-2 Genomes for Molecular Epidemiology across the COVID-19 Pandemic. Viruses, 15(2), 560. https://doi.org/10.3390/v15020560