Abstract
The study aimed to characterize the genotype and subgenotypes of HBV circulating in Saudi Arabia, the presence of clinically relevant mutations possibly associated with resistance to antivirals or immune escape phenomena, and the possible impact of mutations in the structural characteristics of HBV polymerase. Plasma samples from 12 Saudi Arabian HBV-infected patients were analyzed using an in-house PCR method and direct sequencing. Saudi patients were infected with mainly subgenotype D1. A number of mutations in the RT gene (correlated to antiviral resistance) and within and outside the major hydrophilic region of the S gene (claimed to influence immunogenicity and be related to immune escape) were observed in almost all patients. Furthermore, the presence of mutations in the S region caused a change in the tertiary structure of the protein compared with the consensus region. Clinical manifestations of HBV infection may change dramatically as a result of viral and host factors: the study of mutations and protein-associated cofactors might define possible aspects relevant for the natural and therapeutic history of HBV infection.
1. Introduction
Human hepatitis B virus (HBV) is an enveloped DNA virus belonging to the Hepadnaviridae family [1] associated with a wide spectrum of clinical manifestations resulting in both acute and chronic liver infection [2]; in addition, chronic HBV hepatitis can be complicated by liver cirrhosis, and hepatocellular carcinoma [3,4]. Despite the wide implementation of immunization programs, HBV infection still remains a major public health problem with approximately 296 million people who are chronically infected worldwide [5,6,7].
The incidence rate of hepatitis B in Saudi Arabia has declined from 19.65 in 2003 to 13.63 per 100,000 inhabitants in 2016 [8]. Furthermore, a recent cross-sectional study involving 74,662 participants reported a prevalence rate of hepatitis B infection of approximately 1.3% [8]. Nevertheless, hepatitis B continues to be a serious health problem in Saudi Arabia [9], mainly affecting older people with advanced forms of liver disease and several co-morbidities [10]. HBV, despite being a DNA virus, is characterized by a high genetic heterogeneity due to the peculiar mechanism of viral replication requiring the activity of reverse transcriptase (RT) polymerase for the reverse transcription of an RNA intermediate, called pregenomic RNA (pgRNA) [11].
Consequently, there are 10 genotypes of HBV(A–J) until now and several subgenotypes [12]. The predominant genotype in the Eastern Mediterranean countries including Saudi Arabia is genotype D.
Additionally, HBV has a quasispecies distribution in infected individuals; mutations may occur in all of the genes and can be responsible for treatment resistance, immune escape, disease outcome, and carcinogenesis [13,14,15,16].
Mutations in the reverse transcriptase (RT) region of HBV polymerase can reduce the susceptibility to antiviral drugs or the restoration of replicative fitness [17]. In addition, HBV surface antigen (HBsAg) contains the major hydrophilic region (MHR), a dominant epitope crucial for binding to neutralizing antibodies. The presence of mutations in this region might change the hydrophilicity, electric charge, or acidity of the loop, having a number of pathobiological effects on the structure of HBV polymerase. Moreover, due to overlapping between the RT gene and HBsAg, the presence of mutations can also contribute to reduce the binding affinity for neutralizing antibodies, including those induced by HBV vaccine [11].
This study aimed to assess the HBV genotypes and subgenotypes circulating in Saudi Arabia and to investigate the presence of clinically relevant mutations in RT and S region in order to verify the possible impact of mutations on the structural characteristics of HBV polymerase in Saudi infected patients.
2. Patients, Material, and Methods
2.1. Study Population
In this study, we included hepatitis-B-infected patients older than 18 years attending the Hepatology Clinic in the King Fahad Hospital of the University-Al-Khobar (Saudi Arabia) enrolled in the period January to March 2018. The patients were included in the study, after signed informed consent, if they were reactive for HBsAg for more than six months and if they had detectable plasma HBV DNA, while we excluded patients with undetectable plasma HBV DNA, co-infection with HCV or HDV, autoimmune liver disease, primary biliary cirrhosis, hemochromatosis, alpha one antitrypsin deficiency, Wilson’s disease, liver cirrhosis, or hepatic injury caused by drug use; pregnant women and nursing mothers were excluded as well.
The clinical and demographical data were obtained from the patient’s medical records and included: age, sex, and history of previous use of antiviral treatment. The standard clinical investigations included liver function tests (ALT, alanine aminotransferase; AST, aspartate aminotransferase; ALP, alkaline phosphatase; total bilirubin; albumin); platelet count; international normalized ratio (INR); and results of HBV serological markers (HBsAg, HBeAg, anti-HBs, anti-HBc, and anti-HBe) using the Abbott Architect assay (Abbott Diagnostics Division Max-Planck-ring 2, 65205, Wiesbaden, Germany).
Transient elastography (TE) (FibroScan® 502 Touch, Echosens, Paris, France) was used to assess liver fibrosis (liver stiffness measurement), expressed in kpascal (kPa) in combination with controlled attenuation parameter (CAP), used to determine liver steatosis, expressed in decibels per meter (dB/m).
All TE procedures were performed by an experienced practitioner (M.I.) after at least 4 h of fasting by the patients. The measurements were made on the right lobe of the liver, as described by the manufacturing company, and ten successful measurements were obtained for each patient. TE failure was recorded when no value was obtained after at least ten attempts. The results were considered unreliable if the number of valid attempts was fewer than 10, the success rate was <60%, or the interquartile range/median was >30%.
2.2. Plasma Samples
The plasma samples were stored at −80 °C until testing. Plasma samples were obtained from 26 Saudi Arabian HBV-infected patients.
The real-time HBV PCR viral load was performed using the Artus® HBV RG PCR kit (QIAGEN GmbH, QIAGEN Strasse 1, 40724 Hilden, Germany). The lower detection limit was 10 IU/mL.
2.3. HBV Sequencing and Genotyping
HBV genotype and detection of mutations in the polymerase gene were carried out using an in-house PCR method, as shown elsewhere [18]. For each patient, HBV reverse transcriptase (HBV-RT) (344 amino acids) and the overlapped HBsAg (226 amino acids) full-length sequencing was performed on plasma samples, as described previously [18,19].
Briefly, HBV DNA was extracted from 140 uL of plasma using a commercially available kit (QIAmp DNA blood mini kit, Qiagen Inc., 19300 Germantown Rd., Germantown, MD 20874, USA); the first round of PCR was carried out in the final volume of 50 uL containing Ampli Taq Gold polymerase enzyme and using the following primer pairs: 5-GGTCACCATATTCTTGGGAA-3′ and 5′-GTGGGGGTTGCGTCAGCAAA-3′. PCR conditions were: one cycle at 93 °C for 12 min, 40 cycles (94 °C 50 s, 53 °C 50 s, 72 °C 1 min and 30 s), and a final cycle at 72 °C for 10 min. [18]. When the first amplification round provided negative results, a second round of PCR was used. The second round of PCR was carried out using 5 μL of the first PCR product under the same condition as the first round of PCR. The PCR products were electrophoresed on 2% agarose gel and stained with ethidium bromide [18].
The lower detection limit of the described nested PCR in this study was estimated to be 20 copies/mL HBV DNA.
PCR products were sequenced by using eight different overlapping sequence-specific primers with a Big Dye terminator v. 3.1 cycle sequencing kit (Applied Biosystems, Foster City, CA, USA) with the Sanger sequencing method, and an automated sequencer (ABI-3100), as reported elsewhere [18,19]. The sequences were analyzed using Seqscape-v.2.0 software. Amino acid (aa) polymorphisms associated with drug resistance were obtained using geno2pheno HBV. Genotyping was carried out by phylogenetic comparison of all patient sequences (RT and S genomic regions separately) with genotype reference sequences, as recommended by Schaefer [20], that used one reference sequence per subgenotype, labelled in the trees as “GT-X.accession number”, plus a non-human primate virus as out group; 7 + 1 sequences. In addition, BLASTN identified the closest previously published sequences in GenBank that were added to phylogeny; sometimes, the same GenBank sequence was identified as closest to >1 of our sequences. Thus, this resulted in addition 11 and 7 GenBank sequences to the S and RT trees. We did use the SH-test in PhyML to assess reconstruction robustness. All GenBank sequences were subgenotyped as genotype D1 in agreement with their original classification, providing adequate subgenotyping robustness. Sequences were aligned using MAFFT V7 under the G-INS-1 algorithm [21], and phylogenetic trees were calculated using PhyML V3 under a GTR + I + G model and best of NNI + SPR search [22] (Figure 1 and Figure 2).
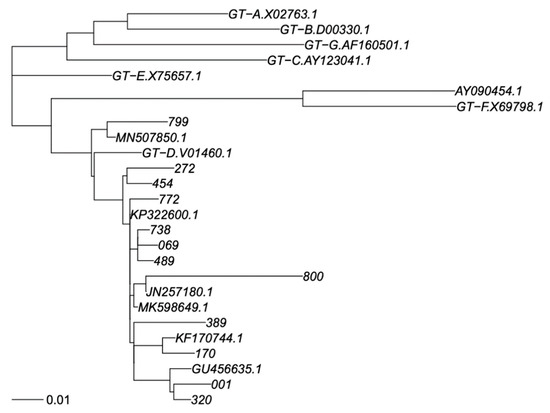
Figure 1.
Phylogenetic tree of RT sequence fragment. Legend: Phylogenetic tree for genotype classification of RT segment. Genotyping reference sequences are labelled “GT-X”, where X indicates genotype, followed by the GenBank accession number. Our new sequences are labelled with a three-digit numerical code, and the closest sequences in GenBank are labelled with their accession number. The scalebar is in units of substitutions/site. Trees were constructed as described in Materials and Methods.
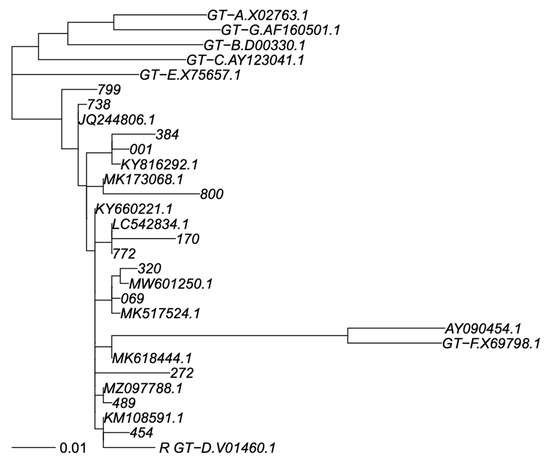
Figure 2.
Phylogenetic tree of S sequence fragment. Legend: Phylogenetic tree for genotype classification of S segment. Genotyping reference sequences are labelled “GT-X”, where X indicates genotype, followed by the GenBank accession number. Our new sequences are labelled with a three-digit numerical code, and the closest sequences in GenBank are labelled with their accession number. The scalebar is in units of substitutions/site. Trees were constructed as described in Materials and Methods.
The online ExPASy ProtParam tool [23], available at http://expasy.org/tools/protparam.html, (accessed on 2 September 2022) was used to study, in both the S gene and the RT gene, molecular weight, theoretical isoelectric point (pI), extinction coefficient, aliphatic index, instability index, grand average of hydropathy (GRAVY), and the total number of positive and negative residual amino acids [23].
The online I-TASSER (I-TASSER Suite 5.2Department of Computational Medicine and Bioinformatics, Department of Biological Chemistry, University of Michigan Medical School, 100 Washtenaw Avenue, Ann Arbor, MI 48109-2218, USA) server was used for automated protein-structure prediction and structure-based function annotation [24,25,26].
3. Results
Demographic, virological, and clinical characteristics of the twenty-six HBV-infected Saudi Arabian patients enrolled in the study are shown in Table 1. Patients were predominantly males (17 were male and 9 were female), with a mean age of 45.2 years (range 31–57). All patients were anti-HBe positive with HBV DNA levels ranging from 1 × 103 to 6 × 106 IU/mL. Liver fibrosis was generally mild (F1–F2), and only three patients were treated with antiretrovirals (tenofovir disoproxil fumarate) (Table 1).

Table 1.
Demographic, virological, and clinical characteristics of patients infected with HBV.
Although, at the time of enrollment in the study, all tested patients had detectable HBV-DNA in plasma, for molecular investigation, only plasma samples obtained from 12 of the patients were suitable for HBV DNA extraction and amplification. For the remaining samples, the absence of HBV DNA was confirmed even after running the second round of PCR amplification.
The phylogenetic tree analysis of the HBV polymerase gene showed that all of the 12 subjects were infected with an HBV D genotype. In particular, we observed that eleven isolates belonged to subgenotype D1 (92%), and one to subgenotype D2 (8%). No deletion or insertion were detected in the polymerase region. However, several amino acid (aa) substitutions were observed in the RT region of HBV polymerase and in the S gene (Table 2 and Table 3).
Table 2.
Distribution of polymorphisms found for RT of HBV polymerase gene.
Table 3.
Distribution of polymorphisms found for the surface gene of HBV.
In the RT gene, the Y135S substitution was observed in 11/12 HBV strains, followed by the N248H substitution found in 10/12 HBV isolates regardless of the HBV subgenotype.
The change of serine to threonine at position 213 was identified in two HBV isolates (Table 2). Only in one HBV isolate (patient 772), we identified a mutation at position 181 (A181G), known to confer resistance to adefovir and tenofovir as well as telbivudine and lamivudine [27,28,29].
Additionally, this strain also showed a mutation at position 233 (I233M) known to be associated with resistance to adefovir, although this resistance included a valine as aa change [30]. The mutation at position 215 associated with resistance to adefovir was observed in 5/12 HBV strains, although two HBV strains showed a modified glutamine in serine, two in histidine, and one in proline. Interestingly, all patients were naïve to the treatment. Other substitutions were also seen at positions 54, 122, 266, 329, 336, and 337 (Table 2).
The presence of aa substitutions at different positions in the surface region was observed in all HBV isolates. At position 207, different aa changes were found in 6/12 HBV strains; the substitutions T118A and L209V were observed in two different HBV isolates (Table 3) and the isolate (HBV_800 strain) showed several aa mutations at positions 129, 131, 133, 193, 203, 204, 205, and 209, including those associated with hepatitis B surface antigen escape. Our study showed mutations within the major hydrophilic region (MHR) of HBsAg in the second loop of “a” determinant associated with escape from vaccine-induced immunity in several HBV isolates (Table 3). A mutation at position 129 (Q129H) was observed in three isolates and additional mutations T131N + M133T were found in the HBV_800 strain, whereas the HBV_272 strain showed more additional mutations at position 109, 120, 126, and 131. The HBV_738 isolate displayed a mutation at position 144 (D144E).
The presence of polymorphisms in the region of the S protein outside the MHR (aa 78-aa 99) associated with antigenicity prediction was observed in 4/12 (33%) of HBV Saudi strains (001; 800; 170; 389).
The physicochemical properties of both polymerase and S genes of HBV isolates are reported in Table 4 and Table 5, as observed by the online ExPASy ProtParam tool (SIB Swiss Institute of Bioinformatics, Quartier Sorge—Batiment Amphipole, 1015 Lausanne, Switzerland [23]. The theoretical isoelectric point (pI) in the polymerase protein was found to be either acidic or neutral in all samples ranging from 5.11 to 7.39 (Table 4).
Table 4.
Molecular weight, theoretical isoelectric point (pI), extinction coefficient, aliphatic index, instability index, grand average of hydropathy (GRAVY), and total number of positive and negative residues of RT of the polymerase region.
Table 5.
Molecular weight, theoretical isoelectric point (pI), extinction coefficient, aliphatic index, instability index, grand average of hydropathy (GRAVY), and total number of positive and negative residues of S protein.
The GRAVY score was indeed similar for all polymerase proteins except one; in fact, 11/12 had proteins with a GRAVY score above 0, whereas, in an HBV isolate, the GRAVY score was below 0 (−0.034) which is considered a hydrophilic protein (globular protein). Our sequences of the polymerase gene with an instability index of more than 44.55 resulted in unstable proteins.
For the S gene, our sequences were identified as alkaline proteins in 11/12 HBV isolates with a pI value above 8.2. However, for the HBV_272 isolate, the pI was not computed because the sequence had multiple polymorphisms. The GRAVY index was greater than 0 in all S proteins, showing a more probable membraneous protein (hydrophobic protein). The instability indices around 51.98–64.12 identified all S proteins as unstable (Table 5).
Tertiary structures of HBsAg from HBV consensus and from HBV_800 (an isolate that showed several mutations) were predicted by I-Tasser and further by DeepFold models (Figure 3, Figure 4 and Figure 5).

Figure 3.
703029 (HBV Consensus) tertiary structure using I-Tasser online software. Pink is alpha-helix; Blue is coil and white region is extended strand.
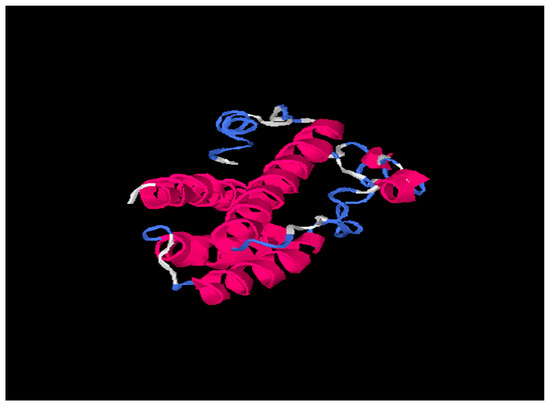
Figure 4.
701347 (HBV_800 isolate) tertiary structure using I-Tasser online software. Pink is alpha-helix; Blue is coil and white region is extended strand.
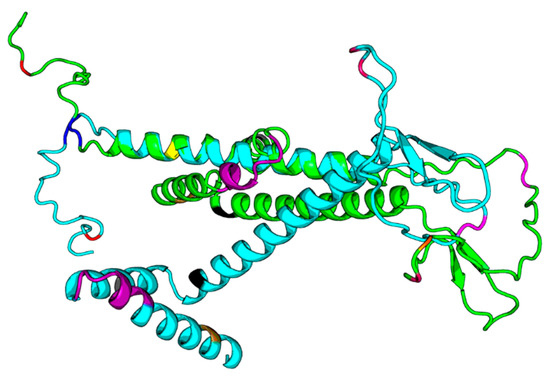
Figure 5.
DF292. DF293.png. Legend: The green structure in the png file is DF292 (HBV consensus), and the cyan one is DF293 (HBV_800 isolate), and we highlight the sequence differences in the structure model with different colors. The two models both have N-terminus helix regions and C-terminus helix regions; however, the orientations between those two helix regions of the two models are quite different.
Our results based on DeepFold models showed that the two proteins seemed to have completely different folds with TM-score = 0.38. The two models both have N-terminus helix region, C-terminus helix region, and a centrally beta-like core region; however, the orientations between those two helix regions of the two models are quite different, mainly due to the mutation on core region changing the fold orientation of the N terminus region and C terminus region (Figure 3).
4. Discussion
Although HBV is a DNA virus, it is characterized by a great variability, with different genotypes and subgenotypes and also an intraindividual variability, probably influencing several aspects of the infection, including prognosis, evolution, response/resistance to antivirals, and sensitivity to natural or vaccine/induced neutralizing antibodies
In this study, we characterized the genotypes and subgenotypes of HBV circulating in Saudi Arabia, the presence of clinically relevant mutations possibly associated with resistance to antivirals or immune escape phenomena, and the possible impact of mutations in the structural characteristics of HBV polymerase.
Phylogenetic analysis showed that all Saudi subjects were infected with HBV genotype D in line with other molecular studies in Saudi Arabia [31,32,33,34] This finding corresponds to the epidemiological profile observed in other neighboring countries [35,36]. Interestingly, in fact, the D1 subgenotype is the dominant subtype in the Mediterranean area [32], whereas the presence of D2 subgenotypes had been found in different geographic areas as Europe, India, Australia, and Africa [37,38,39,40].
In our study, 11 of the HBV isolates studied belonged to the subgenotype D1 (92%) and one to the subgenotype D2 (8%). These results support the subgenotype profile already presented in the recent study using a limited number of isolates from Saudi Arabia [34].
In this paper, different mutations in the RT region of the polymerase peptide as well as in the surface region were observed in all of the HBV Saudi strains.
Two different mutations, Y135S and N248H, in the reverse transcriptase region of the polymerase peptide have been identified among our HBV strains causing infection in the Saudi population. Tuteja and collaborators showed both these two mutations in the HBV strains with the D genotype circulating in the infected Indian population, and the frequency was 67 vs. 59, respectively [29]. We found a mutation at position 248 in RT in 83% of Saudi HBV isolates, independently of D subgenotypes. This is consistent with a study by Chavan and collaborators, in which this mutation was described as the most common genotypic variant in their West Indian population [41].
The substitution in the RT region of serine with threonine at position 213 (S213T) was observed and detected in naïve patients with subgenotype D1. The S213T in the RT region of the polymerase gene was reported to be associated with HBV A2, B, and C genotypes in previous studies [42,43]; moreover, Zhang and colleagues [43] described the S213T mutation and classified it as an unconventional mutation since it is found in patients with virological breakthrough and treated with ADV, ETV, and LMV.
The 772_HBV isolate, from an infected individual naïve to antiviral treatments, exhibited an aa mutation at position 181 (A181G), known to confer resistance to adefovir, tenofovir, telbivudine, and lamivudine [29,44]. Another mutation also associated with adefovir resistance was observed at position 233 (I233M), though this resistance includes valine as an aa modification.
Five out of twelve HBV isolates showed the presence of a mutation at position 215 (Q215S) associated with adefovir resistance, although all these patients were naïve to the treatment.
Sequence analysis of the S gene showed aa substitutions in various positions, including those associated with hepatitis B surface antigen escape [45,46] in several HBV isolates.
Mutations in “a” determinant of surface protein were further observed in 4/12 of HBV isolates of Saudi patients; in previous reports [45,46,47], the presence of mutations in this region correlated with the absence of HBsAg in samples, though in our case we could not confirm this finding.
However, if different mutations or combinations could be involved in the absence /or presence of HBsAg is still unclear.
In our study, the presence of polymorphisms in the region of S protein outside the MHR (78 aa–99 aa) was observed in 4/12 (33%) of Saudi HBV strains (001; 800; 170; 389). It is known that this region might influence the immunogenicity and antigenicity of HBsAg, as reported by Khodadad [47].
The physicochemical properties of HBs protein confirmed the data published by Khodadad [47], showing that all the S proteins were basic, having a pI higher than 8; furthermore, the instability index around 51.98–64.12 identified the S proteins as unstable, and they were recognized as hydrophobic having a GRAVY above 0.
It is known that the clinical manifestations of HBV infection may change radically due to either viral or host factors; therefore, it may be of great importance to study, understand, and define these potentially important co-factors.
Surely, one of the limits of this study was that not all of our HBV samples could be amplified and sequenced. One possible explanation could be that sample storage could have affected DNA extraction/amplification, because of viral DNA degradation.
In conclusion, we characterized in this study the HBV strains circulating in Saudi Arabia, both from the molecular and physicochemical point of view, with the aim of providing new information needed to better clarify the potential outcomes relevant to the natural and therapeutic history of HBV infection.
Author Contributions
M.D.S., J.R.F., T.A.S., T.L. and M.I. planned and supervised the project and the manuscript writing; M.D.S., G.F., T.L., Y.Z. and W.Z. processed samples, performed molecular assays, and supervised the phylogenetic analysis and DeepFold models; M.J.A., M.I. and A.H. collected and supervised the clinical assessment. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Informed Consent Statement
The patients signed a written informed consent in agreement with the WMA Helsinki declaration allowing the use of blood samples for routine clinical analysis to monitor the planned antiviral treatment, including biomolecular studies and the eventual publication of obtained data in an anonymous way.
Data Availability Statement
Data are available on request to the Corresponding Author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rajoriya, N.; Combet, C.; Zoulim, F.; Janssen, H.L. How viral genetic variants and genotypes influence disease and treatment outcome of chronic hepatitis B. Time for an individualised approach? J. Hepatol. 2017, 67, 1281–1297. [Google Scholar] [CrossRef] [PubMed]
- Liaw, Y.-F.; Chu, C.-M. Hepatitis B virus infection. Lancet 2009, 373, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Tarocchi, M.; Polvani, S.; Marroncini, G.; Galli, A. Molecular mechanism of hepatitis B virus-induced hepatocarcinogenesis. World J. Gastroenterol. 2014, 20, 11630–11640. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Han, Q.; Zhao, H.; Zhang, J. The Mechanisms of HBV-Induced Hepatocellular Carcinoma. J. Hepatocell. Carcinoma 2021, ume 8, 435–450. [Google Scholar] [CrossRef]
- World Health Organization. Global Progress Report on HIV, Viral Hepatitis, and Sexually Transmitted Infections. 2021. Available online: https://www.who.int/publications/i/item/9789240027077 (accessed on 2 September 2022).
- Nelson, N.P.; Easterbrook, P.J.; McMahon, B.J. Epidemiology of Hepatitis B Virus Infection and Impact of Vaccination on Disease. Clin. Liver Dis. 2016, 20, 607–628, Erratum in Clin. Liver Dis. 2017, 21, xiii. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Aljumah, A.A.; Babatin, M.; Hashim, A.; Abaalkhail, F.; Bassil, N.; Safwat, M.; Sanai, F.M. Hepatitis B care pathway in Saudi Arabia: Current situation, gaps and actions. Saudi J. Gastroenterol. 2019, 25, 73–80. [Google Scholar] [CrossRef]
- Herzallah, H.K.; Antonisamy, B.R.; Shafee, M.H.; Al-Otaibi, S.T. Temporal trends in the incidence and demographics of cancers, communicable diseases, and non-communicable diseases in Saudi Arabia over the last decade. Saudi Med. J. 2019, 40, 277–286. [Google Scholar] [CrossRef]
- Al-Qahtani, A.A.; Pourkarim, M.R.; Trovão, N.S.; Vergote, V.; Li, G.; Thijssen, M.; Abdo, A.A.; Sanai, F.M.; Cruz, D.D.; Bohol, M.F.F.; et al. Molecular epidemiology, phylogenetic analysis and genotype distribution of hepatitis B virus in Saudi Arabia: Predominance of genotype D1. Infect. Genet. Evol. 2019, 77, 104051. [Google Scholar] [CrossRef]
- Sanai, F.M.; Alghamdi, H.; Alswat, K.A.; Babatin, M.A.; Ismail, M.H.; Alhamoudi, W.K.; Alalwan, A.M.; Dahlan, Y.; Alghamdi, A.S.; Alfaleh, F.Z.; et al. Greater prevalence of comorbidities with increasing age: Cross-sectional analysis of chronic hepatitis B patients in Saudi Arabia. Saudi J. Gastroenterol. 2019, 25, 194–200. [Google Scholar] [CrossRef]
- Torresi, J. The virological and clinical significance of mutations in the overlapping envelope and polymerase genes of hepatitis B virus. J. Clin. Virol. 2002, 25, 97–106. [Google Scholar] [CrossRef]
- Lin, C.-L.; Kao, J.-H. The clinical implications of hepatitis B virus genotype: Recent advances. J. Gastroenterol. Hepatol. 2011, 26 (Suppl. S1), 123–130. [Google Scholar] [CrossRef] [PubMed]
- Caligiuri, P.; Cerruti, R.; Icardi, G.; Bruzzone, B. Overview of hepatitis B virus mutations and their implications in the management of infection. World J. Gastroenterol. 2016, 22, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Lazarevic, I.; Banko, A.; Miljanovic, D.; Cupic, M. Immune-Escape Hepatitis B Virus Mutations Associated with Viral Reactivation upon Immunosuppression. Viruses 2019, 11, 778. [Google Scholar] [CrossRef]
- Inoue, J.; Nakamura, T.; Masamune, A. Roles of Hepatitis B Virus Mutations in the Viral Reactivation after Immunosuppression Therapies. Viruses 2019, 11, 457. [Google Scholar] [CrossRef] [PubMed]
- Leong, J.; Lin, D.; Nguyen, M.H. Hepatitis B surface antigen escape mutations: Indications for initiation of antiviral therapy revisited. World J. Clin. Cases 2016, 4, 71–75. [Google Scholar] [CrossRef]
- Zoulim, F.; Locarnini, S. Hepatitis B Virus Resistance to Nucleos(t)ide Analogues. Gastroenterology 2009, 137, 1593–1608.e2. [Google Scholar] [CrossRef]
- Svicher, V.; Gori, C.; Trignetti, M.; Visca, M.; Micheli, V.; Bernassola, M.; Salpini, R.; Gubertini, G.; Longo, R.; Niero, F.; et al. The profile of mutational clusters associated with lamivudine resistance can be constrained by HBV genotypes. J. Hepatol. 2009, 50, 461–470. [Google Scholar] [CrossRef]
- Salpini, R.; Svicher, V.; Cento, V.; Gori, C.; Bertoli, A.; Scopelliti, F.; Micheli, V.; Cappiello, T.; Spanò, A.; Rizzardini, G.; et al. Characterization of drug-resistance mutations in HBV D-genotype chronically infected patients, naïve to antiviral drugs. Antivir. Res. 2011, 92, 382–385. [Google Scholar] [CrossRef]
- Schaefer, S. Hepatitis B virus taxonomy and hepatitis B virus genotypes. World J. Gastroenterol. 2007, 13, 14–21. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Guindon, S.; Lethiec, F.; Duroux, P.; Gascuel, O. PHYML Online—A web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Res. 2005, 33, W557–W559. [Google Scholar] [CrossRef]
- Roy, S.; Maheshwari, N.; Chauhan, R.; Sen, N.K.; Sharma, A. Structure prediction and functional characterization of secondary metabolite proteins of Ocimum. Bioinformation 2011, 6, 315–319. [Google Scholar] [CrossRef]
- Zheng, W.; Zhang, C.; Li, Y.; Pearce, R.; Bell, E.W.; Zhang, Y. Folding non-homologous proteins by coupling deep-learning contact maps with I-TASSER assembly simulations. Cell Rep. Methods 2021, 1, 100014. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Freddolino, P.L.; Zhang, Y. COFACTOR: Improved protein function prediction by combining structure, sequence and protein–protein interaction information. Nucleic Acids Res. 2017, 45, W291–W299. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Y. I-TASSER server: New development for protein structure and function predictions. Nucleic Acids Res. 2015, 43, W174–W181. [Google Scholar] [CrossRef] [PubMed]
- Santantonio, T.; Fasano, M.; Durantel, S.; Barraud, L.; Heichen, M.; Guastadisegni, A.; Pastore, G.; Zoulim, F. Adefovir dipivoxil resistance patterns in patients with lamivudine-resistant chronic hepatitis B. Antivir. Ther. 2009, 14, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Goyal, M.; Wu, C.H.; Wu, G.Y. The Molecular and Structural Basis of HBV-resistance to Nucleos(t)ide Analogs. J. Clin. Transl. Hepatol. 2014, 2, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Tuteja, A.; Siddiqui, A.B.; Madan, K.; Goyal, R.; Shalimar; Sreenivas, V.; Kaur, N.; Panda, S.K.; Narayanasamy, K.; Subodh, S.; et al. Mutation Profiling of the Hepatitis B Virus Strains Circulating in North Indian Population. PLoS ONE 2014, 9, e91150. [Google Scholar] [CrossRef] [PubMed]
- Schildgen, O.; Olotu, C.; Funk, A.; Zöllner, B.; Helm, M.; Rockstroh, J.K.; Sirma, H. Selection and Counterselection of the rtI233V Adefovir Resistance Mutation during Antiviral Therapy. J. Clin. Microbiol. 2010, 48, 631–634. [Google Scholar] [CrossRef]
- Al Ashgar, H.I.; Imambaccus, H.; Peedikayil, M.C.; Al Thawadi, S.; Al Quaiz, M.; Al Fadda, M.; Al Kahtani, K.; Kagevi, I.; Khan, M.Q. Prevalence of hepatitis B virus genotype in Saudi Arabia: A preliminary report. Indian J. Gastroenterol. 2008, 27, 81–82. [Google Scholar]
- Jamjoom, G.A.; El-Daly, M.M.; Azhar, E.; Fallatah, H.I.; Akbar, H.O.; Babatin, M.; Alghamdi, A.S.; Dgdgi, M.I.; Hamid, M.A.; Qari, Y.A.; et al. Prevalence and molecular characterization of hepatitis D virus in Saudi Arabia: A single-center study. Saudi J. Gastroenterol. 2017, 23, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Al Balwi, M.A.; Tanaka, Y.; Hajeer, A.; Sanai, F.M.; Al Abdulkarim, I.; Al Ayyar, L.; Badri, M.; Saudi, D.; Tamimi, W.; et al. Novel point mutations and mutational complexes in the enhancer II, core promoter and precore regions of hepatitis B virus genotype D1 associated with hepatocellular carcinoma in Saudi Arabia. Int. J. Cancer 2013, 133, 2864–2871. [Google Scholar] [CrossRef] [PubMed]
- EL Hadad, S.; Alakilli, S.; Rabah, S.; Sabir, J. Sequence analysis of sub-genotype D hepatitis B surface antigens isolated from Jeddah, Saudi Arabia. Saudi J. Biol. Sci. 2018, 25, 838–847. [Google Scholar] [CrossRef]
- Alfaresi, M.; Elkoush, A.; Alshehhi, H.; Alzaabi, A.; Islam, A. Hepatitis B virus genotypes and precore and core mutants in UAE patients. Virol. J. 2010, 7, 160. [Google Scholar] [CrossRef] [PubMed]
- Gasim, G.I. Hepatitis B virus in the Arab world: Where do we stand? Arab. J. Gastroenterol. 2013, 14, 35–43. [Google Scholar] [CrossRef]
- Zehender, G.; Ebranati, E.; Gabanelli, E.; Shkjezi, R.; Lai, A.; Sorrentino, C.; Presti, A.L.; Basho, M.; Bruno, R.; Tanzi, E.; et al. Spatial and Temporal Dynamics of Hepatitis B Virus D Genotype in Europe and the Mediterranean Basin. PLoS ONE 2012, 7, e37198. [Google Scholar] [CrossRef]
- Schaefer, S. Hepatitis B virus: Significance of genotypes. J. Viral Hepat. 2005, 12, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Kurbanov, F.; Datta, S.; Chandra, P.K.; Tanaka, Y.; Mizokami, M.; Chakravarty, R. Phylogenetic relatedness and genetic diversity of hepatitis B virus isolates in Eastern India. J. Med. Virol. 2006, 78, 1164–1174. [Google Scholar] [CrossRef]
- Tallo, T.; Tefanova, V.; Priimägi, L.; Schmidt, J.; Katargina, O.; Michailov, M.; Mukomolov, S.; Magnius, L.; Norder, H. D2: Major subgenotype of hepatitis B virus in Russia and the Baltic region. J. Gen. Virol. 2008, 89, 1829–1839. [Google Scholar] [CrossRef]
- Chavan, Y.G.; Pawar, S.R.; Wani, M.; Raut, A.D.; Misra, R.N. Hepatitis B virus DNA polymerase gene polymorphism based prediction of genotypes in chronic HBV patients from Western India. Afr. Health Sci. 2017, 17, 762–772. [Google Scholar] [CrossRef]
- Yano, Y.; Azuma, T.; Hayashi, Y. Variations and mutations in the hepatitis B virus genome and their associations with clinical characteristics. World J. Hepatol. 2015, 7, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, X.; Wei, M.; Zhang, C.; Xu, T.; Liu, L.; Xu, Z. Potential resistant mutations within HBV reverse transcriptase sequences in nucleos(t)ide analogues-experienced patients with hepatitis B virus infection. Sci. Rep. 2019, 9, 8078. [Google Scholar] [CrossRef]
- Borroto-Esoda, K.; Miller, M.D.; Arterburn, S. Pooled analysis of amino acid changes in the HBV polymerase in patients from four major adefovir dipivoxil clinical trials. J. Hepatol. 2007, 47, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Özaslan, M.; Özaslan, E.; Barsgan, A.; Koruk, M. Mutations in the S gene region of hepatitis B virus genotype D in Turkish patients. J. Genet. 2007, 86, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.G.; Ueda, K. Investigation of a Novel Hepatitis B Virus Surface Antigen (HBsAg) Escape Mutant Affecting Immunogenicity. PLoS ONE 2017, 12, e0167871. [Google Scholar] [CrossRef]
- Khodadad, N.; Seyedian, S.S.; Moattari, A.; Haghighi, S.B.; Pirmoradi, R.; Abbasi, S.; Makvandi, M. In silico functional and structural characterization of hepatitis B virus PreS/S-gene in Iranian patients infected with chronic hepatitis B virus genotype D. Heliyon 2020, 6, e04332. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).