
Characterisation of the Upper Respiratory Tract Virome of Feedlot Cattle and Its Association with Bovine Respiratory Disease


 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Viral Metagenomics
2.2. PCR and Sequencing
2.3. Quantitative Real-Time PCR (qPCR)
2.4. Case Control Study
3. Results
3.1. Viral Metagenomics
3.1.1. Orthomyxoviridae
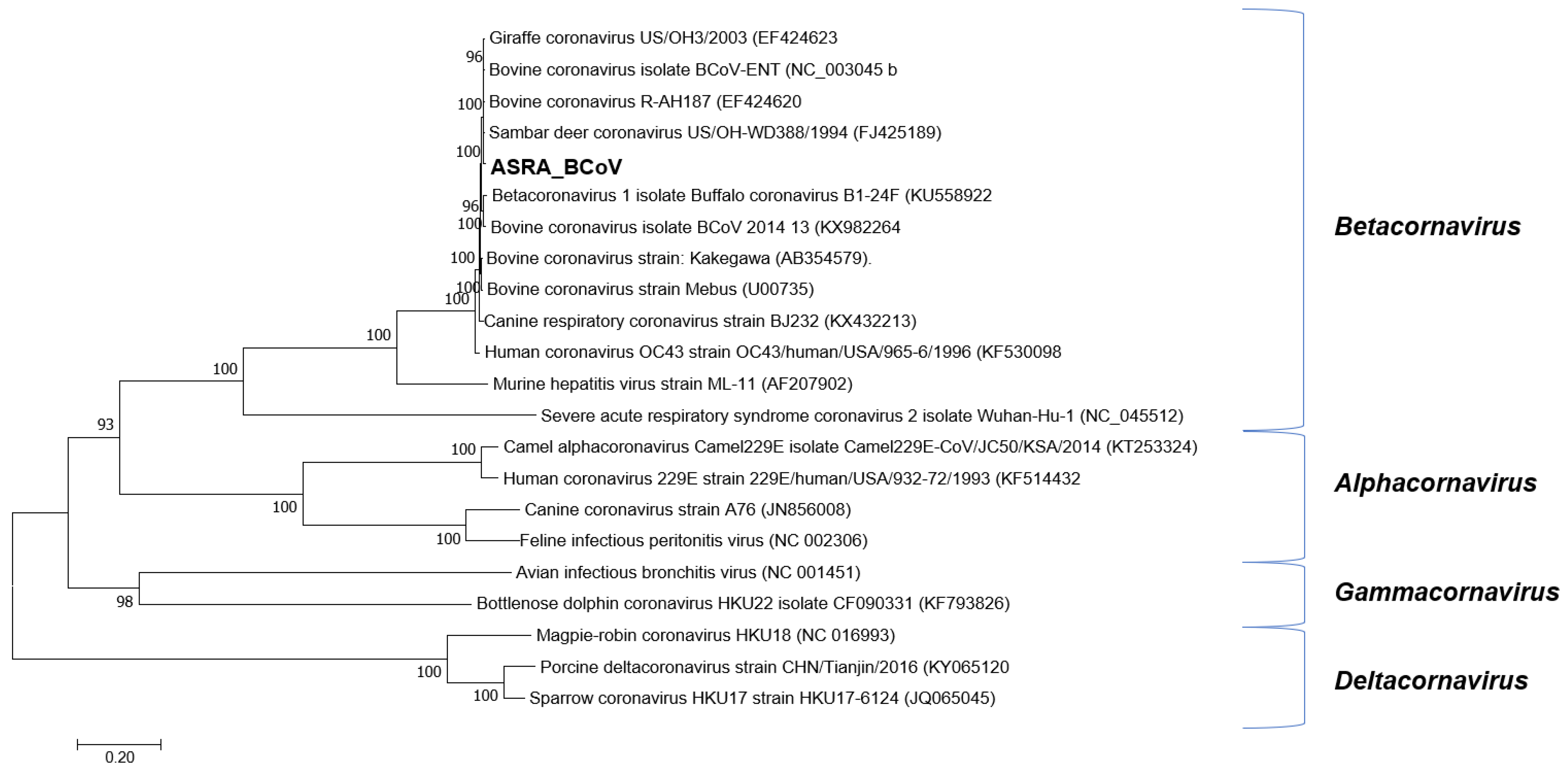
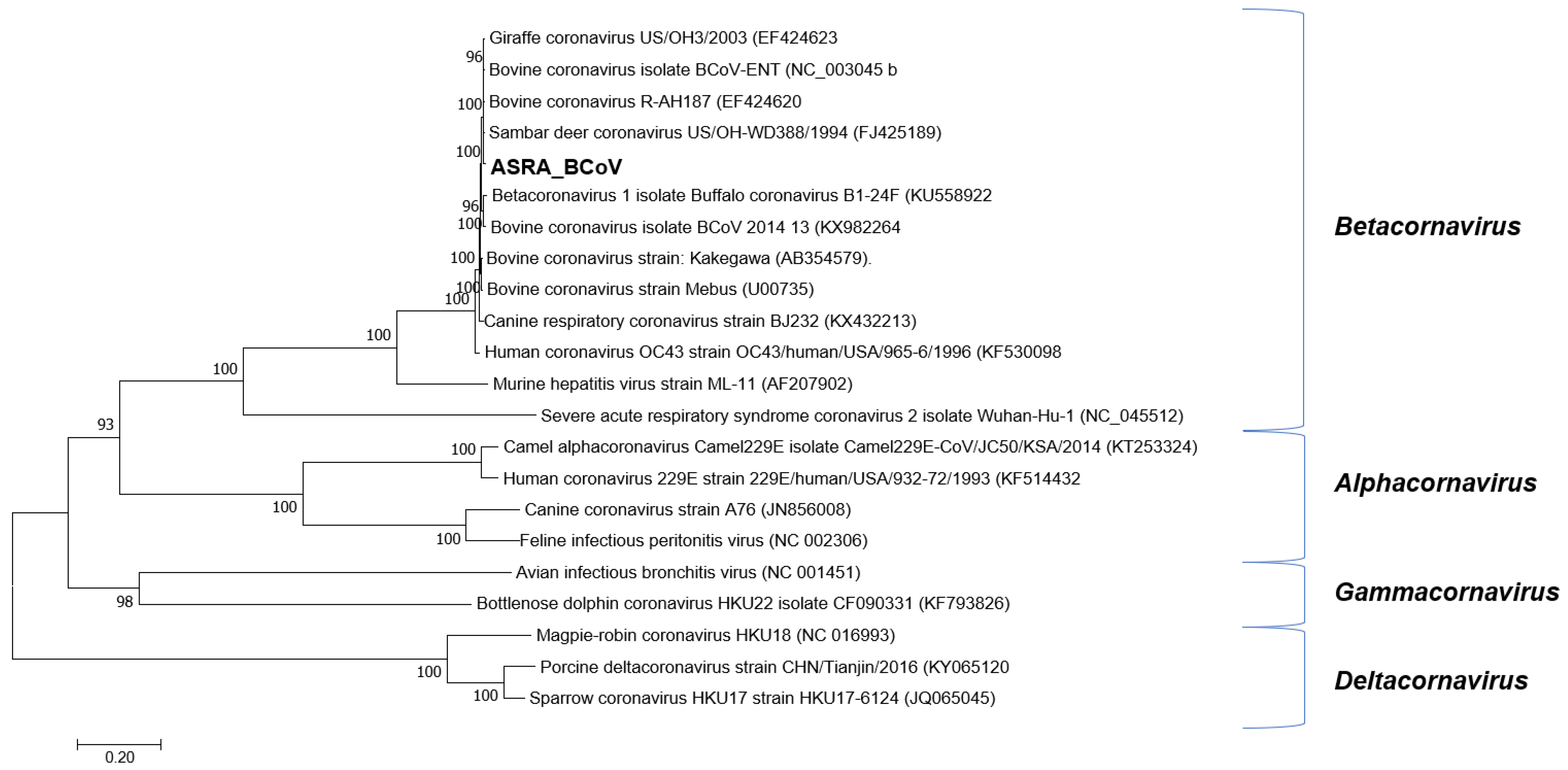
3.1.2. Coronaviridae
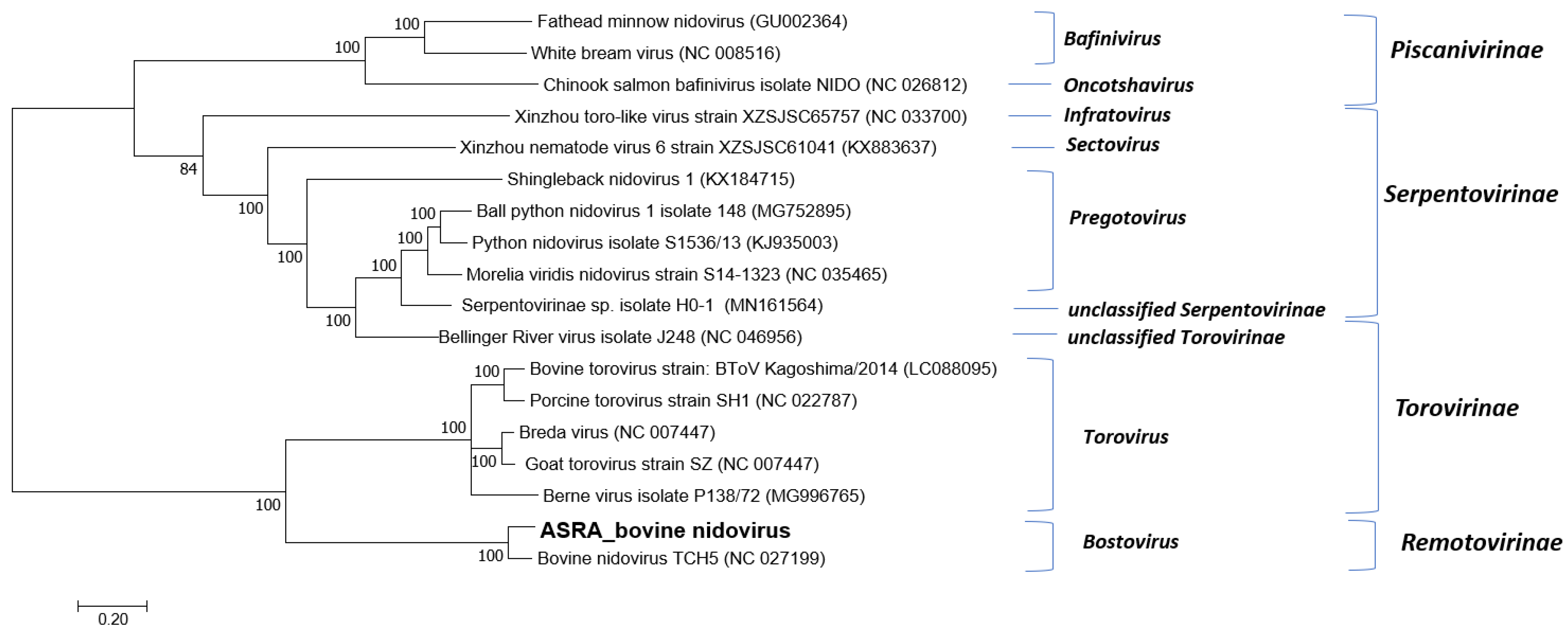
3.1.3. Tobaniviridae
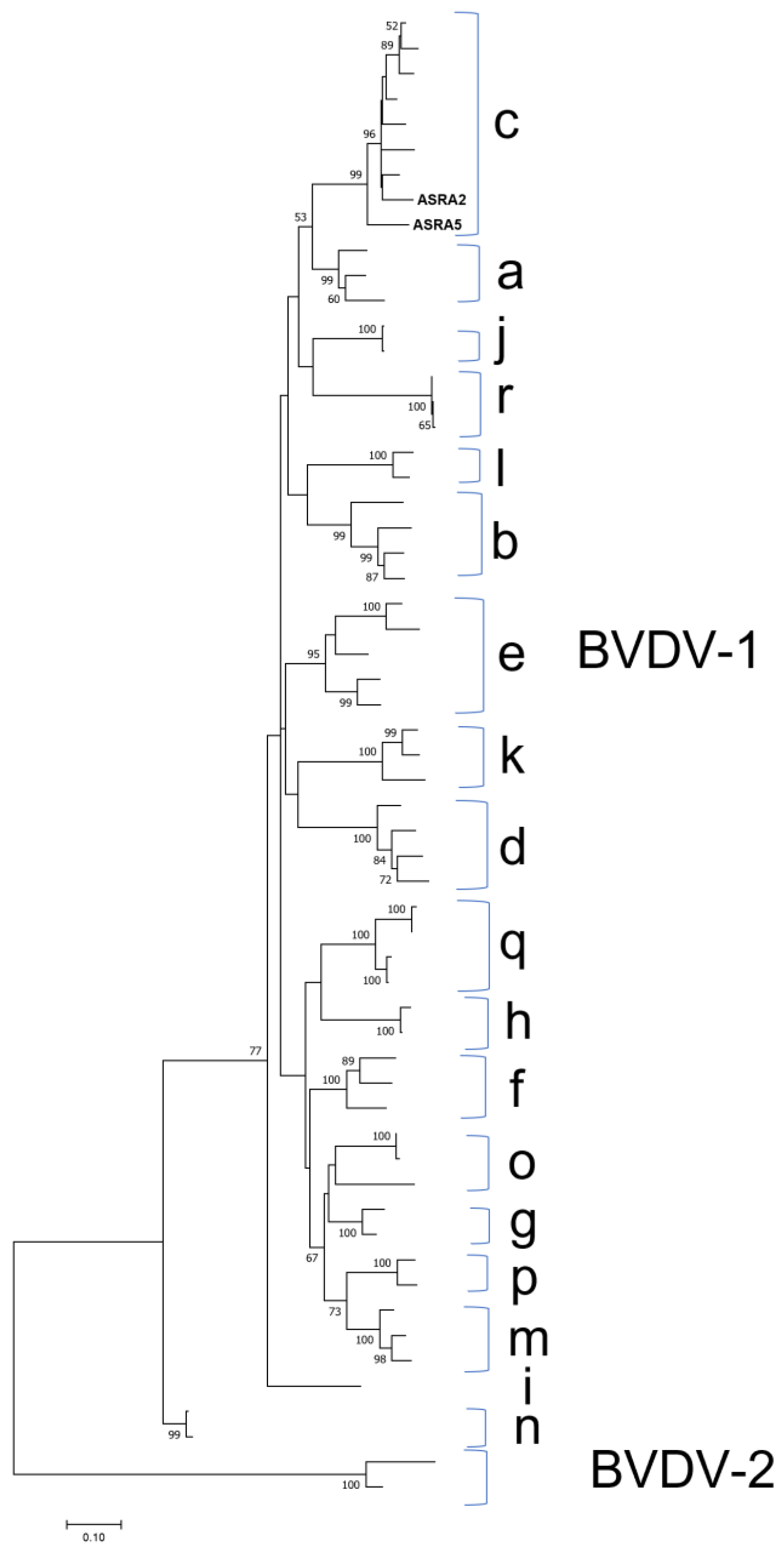
3.1.4. Flaviviridae
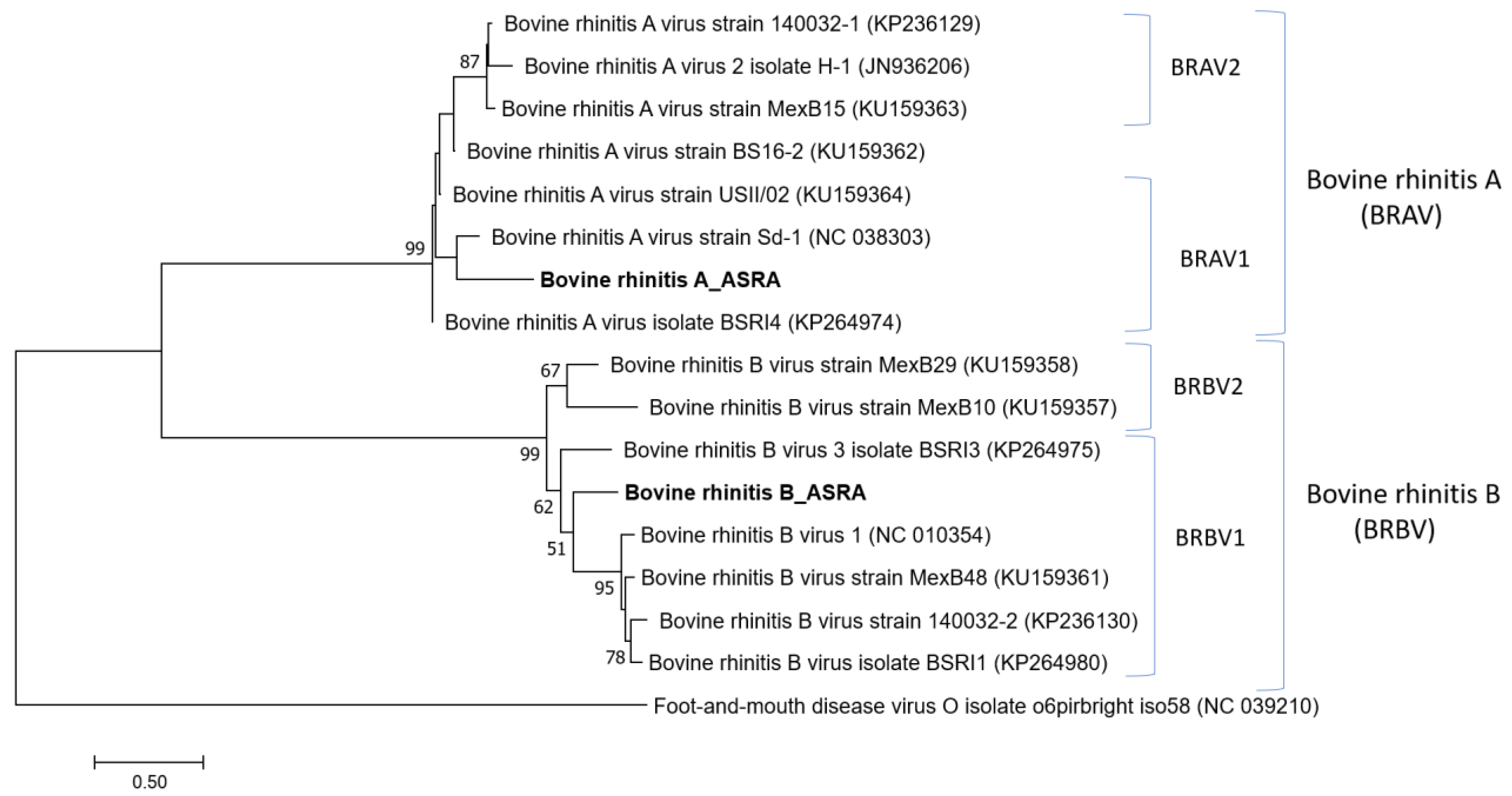
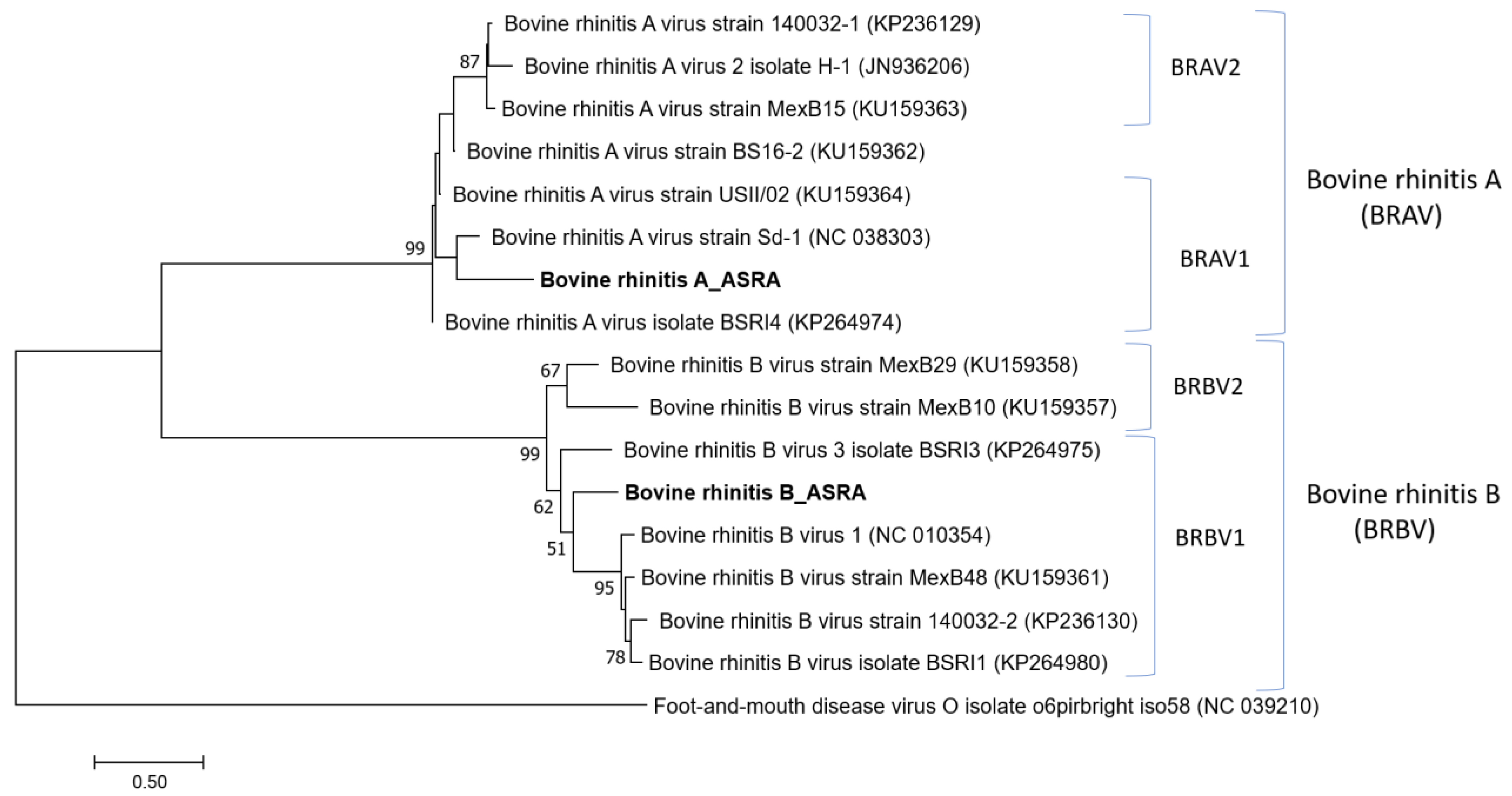
3.1.5. Picornaviridae
3.1.6. Parvoviridae
- Bocoparvovirus
- Erythroparvovirus
- Copiparvovirus
3.1.7. Polyomaviridae
3.1.8. Papillomaviridae
3.2. Case–Control Study of Virus Detection: BRD Cases versus Control Animals
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blakebrough-Hall, C.; McMeniman, J.P.; Gonzalez, L.A. An evaluation of the economic effects of bovine respiratory disease on animal performance, carcass traits, and economic outcomes in feedlot cattle defined using four BRD diagnosis methods. J. Anim. Sci. 2020, 98, skaa005. [Google Scholar] [CrossRef]
- Hay, K.E.; Morton, J.M.; Schibrowski, M.L.; Clements, A.C.; Mahony, T.J.; Barnes, T.S. Associations between prior management of cattle and risk of bovine respiratory disease in feedlot cattle. Prev. Vet. Med. 2016, 127, 37–43. [Google Scholar] [CrossRef]
- Hay, K.E.; Morton, J.M.; Mahony, T.J.; Clements, A.C.; Barnes, T.S. Associations between animal characteristic and environmental risk factors and bovine respiratory disease in Australian feedlot cattle. Prev. Vet. Med. 2016, 125, 66–74. [Google Scholar] [CrossRef]
- Hay, K.E.; Morton, J.M.; Clements, A.C.A.; Mahony, T.J.; Barnes, T.S. Population-level effects of risk factors for bovine respiratory disease in Australian feedlot cattle. Prev. Vet. Med. 2017, 140, 78–86. [Google Scholar] [CrossRef]
- Hay, K.E.; Morton, J.M.; Clements, A.C.; Mahony, T.J.; Barnes, T.S. Associations between feedlot management practices and bovine respiratory disease in Australian feedlot cattle. Prev. Vet. Med. 2016, 128, 23–32. [Google Scholar] [CrossRef]
- Hay, K.E.; Barnes, T.S.; Morton, J.M.; Gravel, J.L.; Commins, M.A.; Horwood, P.F.; Ambrose, R.C.; Clements, A.C.; Mahony, T.J. Associations between exposure to viruses and bovine respiratory disease in Australian feedlot cattle. Prev. Vet. Med. 2016, 127, 121–133. [Google Scholar] [CrossRef]
- Hay, K.E.; Barnes, T.S.; Morton, J.M.; Clements, A.C.; Mahony, T.J. Risk factors for bovine respiratory disease in australian feedlot cattle: Use of a causal diagram-informed approach to estimate effects of animal mixing and movements before feedlot entry. Prev. Vet. Med. 2014, 117, 160–169. [Google Scholar] [CrossRef]
- Hay, K.E.; Ambrose, R.C.; Morton, J.M.; Horwood, P.F.; Gravel, J.L.; Waldron, S.; Commins, M.A.; Fowler, E.V.; Clements, A.C.; Barnes, T.S.; et al. Effects of exposure to bovine viral diarrhoea virus 1 on risk of bovine respiratory disease in Australian feedlot cattle. Prev. Vet. Med. 2016, 126, 159–169. [Google Scholar] [CrossRef]
- Cusack, P.M.; McMeniman, N.P.; Lean, I.J. Effects of injectable vitamins a, d, e and c on the health and growth rate of feedlot cattle destined for the Australian domestic market. Aust. Vet. J. 2008, 86, 81–87. [Google Scholar] [CrossRef]
- Srikumaran, S.; Kelling, C.L.; Ambagala, A. Immune evasion by pathogens of bovine respiratory disease complex. Anim. Health Res. Rev. 2007, 8, 215–229. [Google Scholar] [CrossRef]
- Taylor, J.D.; Fulton, R.W.; Dabo, S.M.; Lehenbauer, T.W.; Confer, A.W. Comparison of genotypic and phenotypic characterization methods for Pasteurella multocida isolates from fatal cases of bovine respiratory disease. J. Vet. Diagn. Invest. 2010, 22, 366–375. [Google Scholar] [CrossRef]
- Murray, G.M.; O’Neill, R.G.; Lee, A.M.; McElroy, M.C.; More, S.J.; Monagle, A.; Earley, B.; Cassidy, J.P. The bovine paranasal sinuses: Bacterial flora, epithelial expression of nitric oxide and potential role in the in-herd persistence of respiratory disease pathogens. PLoS ONE 2017, 12, e0173845. [Google Scholar] [CrossRef]
- Horwood, P.F.; Schibrowski, M.I.; Fowler, E.V.; Gibson, J.S.; Barnes, T.S.; Mahony, T.J. Is Mycoplasma bovis a missing component of the bovine respiratory disease complex in australia? Aust. Vet. J. 2014, 92, 185–191. [Google Scholar] [CrossRef]
- Ng, T.F.; Kondov, N.O.; Deng, X.; Van Eenennaam, A.; Neibergs, H.L.; Delwart, E. A metagenomics and case-control study to identify viruses associated with bovine respiratory disease. J. Virol. 2015, 89, 5340–5349. [Google Scholar] [CrossRef]
- Victoria, J.G.; Kapoor, A.; Li, L.; Blinkova, O.; Slikas, B.; Wang, C.; Naeem, A.; Zaidi, S.; Delwart, E. Metagenomic analyses of viruses in stool samples from children with acute flaccid paralysis. J. Virol. 2009, 83, 4642–4651. [Google Scholar] [CrossRef]
- Freeman, C.N.; Herman, E.K.; Abi Younes, J.; Ramsay, D.E.; Erikson, N.; Stothard, P.; Links, M.G.; Otto, S.J.G.; Waldner, C. Evaluating the potential of third generation metagenomic sequencing for the detection of BRD pathogens and genetic determinants of antimicrobial resistance in chronically ill feedlot cattle. BMC Vet. Res. 2022, 18, 211. [Google Scholar] [CrossRef]
- Zou, S.; Caler, L.; Colombini-Hatch, S.; Glynn, S.; Srinivas, P. Research on the human virome: Where are we and what is next. Microbiome 2016, 4, 32. [Google Scholar] [CrossRef]
- Mitra, N.; Cernicchiaro, N.; Torres, S.; Li, F.; Hause, B.M. Metagenomic characterization of the virome associated with bovine respiratory disease in feedlot cattle identified novel viruses and suggests an etiologic role for influenza D virus. J. Gen. Virol. 2016, 97, 1771–1784. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome. Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de bruijn graphs. Genome. Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; McEwen, G.K.; Margulies, E.H.; Birney, E. Pebble and rock band: Heuristic resolution of repeats and scaffolding in the velvet short-read de novo assembler. PLoS ONE 2009, 4, e8407. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. Blast+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Cock, P.J.; Chilton, J.M.; Gruning, B.; Johnson, J.E.; Soranzo, N. Ncbi blast+ integrated into galaxy. Gigascience 2015, 4, 39. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. Mega7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Tokarz, R.; Sameroff, S.; Hesse, R.A.; Hause, B.M.; Desai, A.; Jain, K.; Ian Lipkin, W. Discovery of a novel nidovirus in cattle with respiratory disease. J. Gen. Virol. 2015, 96, 2188–2193. [Google Scholar] [CrossRef]
- Hause, B.M.; Collin, E.A.; Anderson, J.; Hesse, R.A.; Anderson, G. Bovine rhinitis viruses are common in U.S. Cattle with bovine respiratory disease. PLoS ONE 2015, 10, e0121998. [Google Scholar] [CrossRef]
- Hause, B.M.; Collin, E.A.; Liu, R.; Huang, B.; Sheng, Z.; Lu, W.; Wang, D.; Nelson, E.A.; Li, F. Characterization of a novel influenza virus in cattle and swine: Proposal for a new genus in the Orthomyxoviridae family. Mbio 2014, 5, e00031-14. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kishimoto, M.; Tsuchiaka, S.; Rahpaya, S.S.; Hasebe, A.; Otsu, K.; Sugimura, S.; Kobayashi, S.; Komatsu, N.; Nagai, M.; Omatsu, T.; et al. Development of a one-run real-time pcr detection system for pathogens associated with bovine respiratory disease complex. J. Vet. Med. Sci. 2017, 79, 517–523. [Google Scholar] [CrossRef]
- Blakebrough-Hall, C.; Hick, P.; Gonzalez, L.A. Predicting bovine respiratory disease outcome in feedlot cattle using latent class analysis. J. Anim. Sci. 2020, 98, skaa381. [Google Scholar] [CrossRef]
- Blakebrough-Hall, C.; Dona, A.; D’Occhio, M.J.; McMeniman, J.; Gonzalez, L.A. Diagnosis of bovine respiratory disease in feedlot cattle using blood (1)h NMR metabolomics. Sci. Rep. 2020, 10, 115. [Google Scholar] [CrossRef]
- Horwood, P.F.; Mahony, T.J. Multiplex real-time RT-PCR detection of three viruses associated with the bovine respiratory disease complex. J. Virol. Methods 2011, 171, 360–363. [Google Scholar] [CrossRef]
- Szumilas, M. Explaining odds ratios. J. Can. Acad. Child Adolesc. Psychiatry 2010, 19, 227–229. [Google Scholar]
- Altman, D.G.; Bland, J.M. How to obtain the p value from a confidence interval. BMJ 2011, 343, d2304. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar]
- Tamura, K. Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G+C-content biases. Mol. Biol. Evol. 1992, 9, 678–687. [Google Scholar]
- Chouljenko, V.N.; Foster, T.P.; Lin, X.; Storz, J.; Kousoulas, K.G. Elucidation of the genomic nucleotide sequence of bovine coronavirus and analysis of cryptic leader mrna fusion sites. Adv. Exp. Med. Biol. 2001, 494, 49–55. [Google Scholar]
- Lau, S.K.; Tsang, A.K.; Shakeel Ahmed, S.; Mahbub Alam, M.; Ahmed, Z.; Wong, P.C.; Yuen, K.Y.; Woo, P.C. First genome sequences of buffalo coronavirus from water buffaloes in bangladesh. New Microbes New Infect. 2016, 11, 54–56. [Google Scholar] [CrossRef] [PubMed]
- Brian, D.A.; Baric, R.S. Coronavirus genome structure and replication. Curr. Top Microbiol. Immunol. 2005, 287, 1–30. [Google Scholar] [PubMed]
- Zhang, J.; Guy, J.S.; Snijder, E.J.; Denniston, D.A.; Timoney, P.J.; Balasuriya, U.B. Genomic characterization of equine coronavirus. Virology 2007, 369, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, R.K.; Gravel, J.L.; Commins, M.A.; Fowler, E.V.; Mahony, T.J. In vivo characterisation of five strains of bovine viral diarrhoea virus 1 (subgenotype 1c). Pathogens 2018, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- Mahony, T.J.; McCarthy, F.M.; Gravel, J.L.; Corney, B.; Young, P.L.; Vilcek, S. Genetic analysis of bovine viral diarrhoea viruses from australia. Vet. Microbiol. 2005, 106, 1–6. [Google Scholar] [CrossRef]
- Cotmore, S.F.; Tattersall, P. Parvoviruses: Small does not mean simple. Annu. Rev. Virol. 2014, 1, 517–537. [Google Scholar] [CrossRef]
- Sadeghi, M.; Kapusinszky, B.; Yugo, D.M.; Phan, T.G.; Deng, X.; Kanevsky, I.; Opriessnig, T.; Woolums, A.R.; Hurley, D.J.; Meng, X.J.; et al. Virome of us bovine calf serum. Biologicals 2017, 46, 64–67. [Google Scholar] [CrossRef]
- Miles, D.G. Overview of the north american beef cattle industry and the incidence of bovine respiratory disease (brd). Anim. Health Res. Rev. 2009, 10, 101–103. [Google Scholar] [CrossRef] [PubMed]
- Edwards, T.A. Control methods for bovine respiratory disease for feedlot cattle. Vet. Clin. N. Am. Food. Anim. Pract. 2010, 26, 273–284. [Google Scholar] [CrossRef]
- Zhang, M.; Hill, J.E.; Fernando, C.; Alexander, T.W.; Timsit, E.; van der Meer, F.; Huang, Y. Respiratory viruses identified in western canadian beef cattle by metagenomic sequencing and their association with bovine respiratory disease. Transbound. Emerg. Dis. 2019, 66, 1379–1386. [Google Scholar] [CrossRef]
- Zhang, M.; Hill, J.E.; Godson, D.L.; Ngeleka, M.; Fernando, C.; Huang, Y. The pulmonary virome, bacteriological and histopathological findings in bovine respiratory disease from western Canada. Transbound. Emerg. Dis. 2020, 67, 924–934. [Google Scholar] [CrossRef]
- Richeson, J.T.; Falkner, T.R. Bovine respiratory disease vaccination: What is the effect of timing? Vet. Clin. N. Am. Food Anim. Pract. 2020, 36, 473–485. [Google Scholar] [CrossRef]
- Wang, M.; Yan, Y.; Wang, R.; Wang, L.; Zhou, H.; Li, Y.; Tang, L.; Xu, Y.; Jiang, Y.; Cui, W.; et al. Simultaneous detection of bovine rotavirus, bovine parvovirus, and bovine viral diarrhea virus using a gold nanoparticle-assisted PCR assay with a dual-priming oligonucleotide system. Front. Microbiol. 2019, 10, 2884. [Google Scholar] [CrossRef]
- Allander, T.; Emerson, S.U.; Engle, R.E.; Purcell, R.H.; Bukh, J. A virus discovery method incorporating dnase treatment and its application to the identification of two bovine parvovirus species. Proc. Natl. Acad. Sci. USA 2001, 98, 11609–11614. [Google Scholar] [CrossRef]
- Jager, M.C.; Tomlinson, J.E.; Lopez-Astacio, R.A.; Parrish, C.R.; Van de Walle, G.R. Small but mighty: Old and new parvoviruses of veterinary significance. Virol. J. 2021, 18, 1–29. [Google Scholar] [CrossRef]
- Liu, R.; Sheng, Z.; Huang, C.; Wang, D.; Li, F. Influenza d virus. Curr. Opin. Virol. 2020, 44, 154–161. [Google Scholar] [CrossRef]
- Ferguson, L.; Eckard, L.; Epperson, W.B.; Long, L.P.; Smith, D.; Huston, C.; Genova, S.; Webby, R.; Wan, X.F. Influenza d virus infection in mississippi beef cattle. Virology 2015, 486, 28–34. [Google Scholar] [CrossRef]
- Nissly, R.H.; Zaman, N.; Ibrahim, P.A.S.; McDaniel, K.; Lim, L.; Kiser, J.N.; Bird, I.; Chothe, S.K.; Bhushan, G.L.; Vandegrift, K.; et al. Influenza c and d viral load in cattle correlates with bovine respiratory disease (BRD): Emerging role of orthomyxoviruses in the pathogenesis of brd. Virology 2020, 551, 10–15. [Google Scholar] [CrossRef]
- Saegerman, C.; Gaudino, M.; Savard, C.; Broes, A.; Ariel, O.; Meyer, G.; Ducatez, M.F. Influenza D virus in respiratory disease in Canadian, province of Quebec, cattle: Relative importance and evidence of new reassortment between different clades. Transbound. Emerg. Dis. 2022, 69, 1227–1245. [Google Scholar] [CrossRef]
- Flynn, O.; Gallagher, C.; Mooney, J.; Irvine, C.; Ducatez, M.; Hause, B.; McGrath, G.; Ryan, E. Influenza d virus in cattle, ireland. Emerg. Infect. Dis. 2018, 24, 389–391. [Google Scholar] [CrossRef]
- Hierweger, M.M.; Koch, M.C.; Seuberlich, T. Bovine polyomavirus 2 is a probable cause of non-suppurative encephalitis in cattle. Pathogens 2020, 9, 620. [Google Scholar] [CrossRef]
- Giannitti, F.; da Silva Silveira, C.; Bullock, H.; Beron, M.; Fernandez-Ciganda, S.; Benitez-Galeano, M.J.; Rodriguez-Osorio, N.; Silva-Flannery, L.; Perdomo, Y.; Cabrera, A.; et al. Bovine polyomavirus-1 (epsilonpolyomavirus bovis): An emerging fetal pathogen of cattle that causes renal lesions resembling polyomavirus-associated nephropathy of humans. Viruses 2022, 14, 2042. [Google Scholar] [CrossRef]
- Hatama, S.; Nishida, T.; Kadota, K.; Uchida, I.; Kanno, T. Bovine papillomavirus type 9 induces epithelial papillomas on the teat skin of heifers. Vet. Microbiol. 2009, 136, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Vu, D.L.; Kaiser, L. The concept of commensal viruses almost 20 years later: Redefining borders in clinical virology. Clin. Microbiol. Infect. 2017, 23, 688–690. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Ma, W.T.; Pang, M.; Fan, Q.L.; Hua, J.L. The commensal microbiota and viral infection: A comprehensive review. Front. Immunol. 2019, 10, 1551. [Google Scholar] [CrossRef] [PubMed]
- Hick, P.M.; Read, A.J.; Lugton, I.; Busfield, F.; Dawood, K.E.; Gabor, L.; Hornitzky, M.; Kirkland, P.D. Coronavirus infection in intensively managed cattle with respiratory disease. Aust. Vet. J. 2012, 90, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Blakebrough-Hall, C.; Hick, P.; Mahony, T.J.; Gonzalez, L.A. Factors associated with bovine respiratory disease case fatality in feedlot cattle. J. Anim. Sci. 2022, 100. [Google Scholar] [CrossRef]
- Smith, G.A.; Young, P.L.; Reed, K.C. Emergence of a new bovine herpesvirus 1 strain in australian feedlots. Arch. Virol. 1995, 140, 599–603. [Google Scholar] [CrossRef]
- Smith, G.A.; Young, P.L.; Mattick, J.S. Bovine herpesvirus 1.1—An exotic disease agent? Aust. Vet. J. 1993, 70, 272–273. [Google Scholar] [CrossRef]
- Horwood, P.F.; Gravel, J.L.; Mahony, T.J. Identification of two distinct bovine parainfluenza virus type 3 genotypes. J. Gen. Virol. 2008, 89, 1643–1648. [Google Scholar] [CrossRef]
- Grellet, E.; L’Hote, I.; Goulet, A.; Imbert, I. Replication of the coronavirus genome: A paradox among positive-strand rna viruses. J. Biol. Chem. 2022, 298, 101923. [Google Scholar] [CrossRef] [PubMed]
- McDaneld, T.G.; Kuehn, L.A.; Keele, J.W. Evaluating the microbiome of two sampling locations in the nasal cavity of cattle with bovine respiratory disease complex (brdc). J. Anim. Sci. 2018, 96, 1281–1287. [Google Scholar] [CrossRef] [PubMed]
- Hoper, D.; Mettenleiter, T.C.; Beer, M. Metagenomic approaches to identifying infectious agents. Rev. Sci. Tech. 2016, 35, 83–93. [Google Scholar] [CrossRef]
- Wylezich, C.; Papa, A.; Beer, M.; Hoper, D. A versatile sample processing workflow for metagenomic pathogen detection. Sci. Rep. 2018, 8, 13108. [Google Scholar] [CrossRef] [PubMed]
- Nagy, A.; Abdallah, F.; El Damaty, H.M.; Tariq, A.; Merwad, A.M.A.; Alhatlani, B.Y.; Elsohaby, I. Genetic characterization of upper respiratory tract virome from nonvaccinated egyptian cow-calf operations. PLoS ONE 2022, 17, e0267036. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Pathogen | Name | Primer/Probe Sequence 5′-3′ 1 |
|---|---|---|
| Bovine nidovirus | BNV_Fwd | GTCAACTGGAGTAGGTCGAAAG |
| BNV_Rev | TCAGCCTCATTCCTAACATCAC | |
| BNV_Probe | TEX615-AGGTACCATTACTATACTGAGCTGGCAGC-BHQ-2 | |
| Bovine rhinitis A virus | BRAV_Fwd | AGGTACCCGGAGGTAACAA |
| BRAV_Rev | GGTGCCTGATGAGACATAGAAG | |
| BRAV_Probe | 6FAM-CCCAGGTCAGATCCAGAGTGTCAC-BHQ-1 | |
| Bovine rhinitis B virus | BRBV_Fwd | GCGATTGTGTCCTAGGGTTT |
| BRBV_Rev | GCCACTGAGGTTAGCTTCTC | |
| BRBV_Probe | Cy5-CTGTCCTTTGCACGGCGTGG-BHQ-2 2 | |
| Influenza D virus | IDV_Fwd | GAGGAATGCTGATGGGAATGT |
| IDV_Reverse | CTTTGTAGCCCAGTCCAGTAAC | |
| IDV_Probe | HEX-ATTACAGGGAGGAAGCATTGGCCA-BHQ-1 | |
| Ungulate bocaparvovirus 6 | UBPV6_Fwd | GGGAAGAGTGGCTTCAGTTTAG |
| UBPV6_Rev | GGCTCTTCTCCTTGTTCTTCTG | |
| UBPV6_Probe | HEX-TCCAGATACAATCAGAAGAAGCGCCA-ZEN/IABkFQ |
| IDV Sequence Data (Current Study) | IDV Reference | |||||||
|---|---|---|---|---|---|---|---|---|
| Segment | Protein | Length (nt) | Coverage (%) | Nucleotide Identity (%) | Amino Acid Similarity (%) | GenBank | Length (nt) | Reference Accession |
| 1 | polymerase PB2 | 1054 | 44.6% | 98.0 | 99.2 | OQ348274 | 2364 | LC270265.1 |
| 2 | polymerase PB1 | 929 | 39.9% | 98.5 | 99.7 | OQ348275 | 2330 | LC270266.1 |
| 3 | polymerase 3 P3 | 918 | 41.0% | 98.4 | 98.7 | OQ348276 | 2195 | LC270267.1 |
| 4 | haemagglutinin-esterase HE | 248 | 12.1% | 94.8 | 92.7 | OQ348277 | 2049 | LC270268.1 |
| 5 | nucleoprotein | NT 1 | 1775 | LC270269.1 | ||||
| 6 | P42 | NT 1 | 1219 | LC270270.1 | ||||
| 7 | non-structural protein 2 | NT 1 | 868 | LC270271.1 | ||||
| Open Reading Frame | BCoV-Aus Bases | BCoV-ENT Bases; Identity (%) | BuCoV B1-28F Bases; Identity (%) |
|---|---|---|---|
| Complete genome | 30,999 | 31,028; 98.9 | 30,985; 98.5 |
| orf 1ab polyprotein | 21,278 | 21,284; 99.1 | 21,284; 98.4 |
| 32 kDa nonstructural protein | 837 | 837; 98.7 | 837; 98.1 |
| haemaglutinin esterase (HE) | 1275 | 1275; 99.1 | 1275; 97.7 |
| spike structural protein (S) | 4092 | 4092; 98.3 | 4092; 98.8 |
| 4.9 kDa nonstructural protein | 89 | 90; 92.2 | 78; 100 |
| 4.8 kDa nonstructural protein | 142 | 138; 85.6 | 135; 90.2 |
| 12.7 kDa nonstructural protein | 330 | 330; 98.5 | 330; 99.4 |
| small membrane protein (E) | 255 | 255; 100 | 255; 99.6 |
| matrix protein (M) | 693 | 693; 98.7 | 693; 100 |
| nucleocapsid protein (N) | 1347 | 1347; 98.7 | 1347; 99.5 |
| internal protein (I) | 624 | 624; 98.7 | 624; 99.5 |
| Characteristic | BNV-Aus | BNV TCH5 | Nucleotide Identity (%) | Amino Acid Similarity (%) |
|---|---|---|---|---|
| complete genome | 20,262 | 20,261 | 85.9 | |
| replicase polyprotein (pp1a/b) | 15,323 | 15,332 | 87.2 | 90.5 |
| glycoprotein S (S) | 1686 | 1689 | 81.9 | 83.5 |
| membrane protein 1 (M1) | 696 | 696 | 87.2 | 91.8 |
| nucleocapsid (N) | 534 | 537 | 85.8 | 86.5 |
| glycoprotein G2 (G2) | 1371 | 1368 | 75.6 | 64.1 |
| hypothetical protein | 267 | 267 | 94.8 | 92.0 |
| Risk Factor | qPCR Result | Cases (%) | Controls (%) | OR | 95%CI | p Value |
|---|---|---|---|---|---|---|
| Infected with one or more virus | Positive | 61 (43.3) | 35 (23.8) | 2.4 | 1.5–4.0 | 0.0005 |
| Negative | 80 (56.7) | 112 (76.2) | ||||
| Bovine herpesvirus 1 | Positive | 19 (13.5) | 0 (0) | 47 | 2.8–785.8 | 0.0074 |
| Negative | 122 (86.5) | 147 (100) | ||||
| Bovine coronavirus | Positive | 2 (1.4) | 3 (2.0) | 0.7 | 0.1–4.2 | 0.7 |
| Negative | 139 (98.6) | 144 (98.0) | ||||
| Bovine respiratory syncytial virus | Positive | 6 (4.3) | 2 (1.4) | 3.2 | 0.6–16.2 | 0.2 |
| Negative | 135 (95.7) | 147 (98.6) | ||||
| Bovine parainfluenza virus | Positive | 1 (0.7) | 0 (0) | 3.1 | 0.1–78 | 0.5 |
| Negative | 140 (99.3) | 147 (100) | ||||
| Bovine viral diarrhoea virus 1 | Positive | 3 (2.1) | 0 (0) | 7.5 | 0.4–145.6 | 0.2 |
| Negative | 138 (97.9) | 147 (100) | ||||
| Influenza D virus | Positive | 16 (11.3) | 10 (6.8) | 1.8 | 0.8–4.0 | 0.2 |
| Negative | 125 (88.7) | 137 (93.2) | ||||
| Bovine rhinitis A virus | Positive | 3 (2.1) | 9 (6.1) | 0.3 | 0.1–1.3 | 0.1 |
| Negative | 138 (97.9) | 138 (93.9) | ||||
| Bovine rhinitis B virus | Positive | 0 (0) | 0 (0) | Not carried out | - | - |
| Negative | 141 (100) | 147(100) | ||||
| Bovine nidovirus | Positive | 15 (10.6) | 14 (9.5) | 1.1 | 0.5–2.4 | 0.8 |
| Negative | 126 (89.4) | 133 (90.5) | ||||
| Ungulate bocaparvovirus 6 | Positive | 8 (5.7) | 2 (1.4) | 4.4 | 0.9–20.9 | 0.07 |
| Negative | 133 (94.3) | 145 (98.6) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ambrose, R.K.; Blakebrough-Hall, C.; Gravel, J.L.; Gonzalez, L.A.; Mahony, T.J. Characterisation of the Upper Respiratory Tract Virome of Feedlot Cattle and Its Association with Bovine Respiratory Disease. Viruses 2023, 15, 455. https://doi.org/10.3390/v15020455
Ambrose RK, Blakebrough-Hall C, Gravel JL, Gonzalez LA, Mahony TJ. Characterisation of the Upper Respiratory Tract Virome of Feedlot Cattle and Its Association with Bovine Respiratory Disease. Viruses. 2023; 15(2):455. https://doi.org/10.3390/v15020455
Chicago/Turabian StyleAmbrose, Rebecca K., Claudia Blakebrough-Hall, Jennifer L. Gravel, Luciano A. Gonzalez, and Timothy J. Mahony. 2023. "Characterisation of the Upper Respiratory Tract Virome of Feedlot Cattle and Its Association with Bovine Respiratory Disease" Viruses 15, no. 2: 455. https://doi.org/10.3390/v15020455
APA StyleAmbrose, R. K., Blakebrough-Hall, C., Gravel, J. L., Gonzalez, L. A., & Mahony, T. J. (2023). Characterisation of the Upper Respiratory Tract Virome of Feedlot Cattle and Its Association with Bovine Respiratory Disease. Viruses, 15(2), 455. https://doi.org/10.3390/v15020455