
I329L: A Dual Action Viral Antagonist of TLR Activation Encoded by the African Swine Fever Virus (ASFV)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Homology Modelling of I329L
2.2. Cell Culture
2.3. Plasmids
2.4. Production and Measurement of Lentivirus
2.5. Immunofluorescence
2.6. Luciferase Reporter Gene Assay
2.7. Immunoprecipitation to Demonstrate Association of I329L and TLR3
2.8. Immunoprecipitation to Demonstrate Association of ICD-I329L and TRIF
2.9. ELISA
3. Results
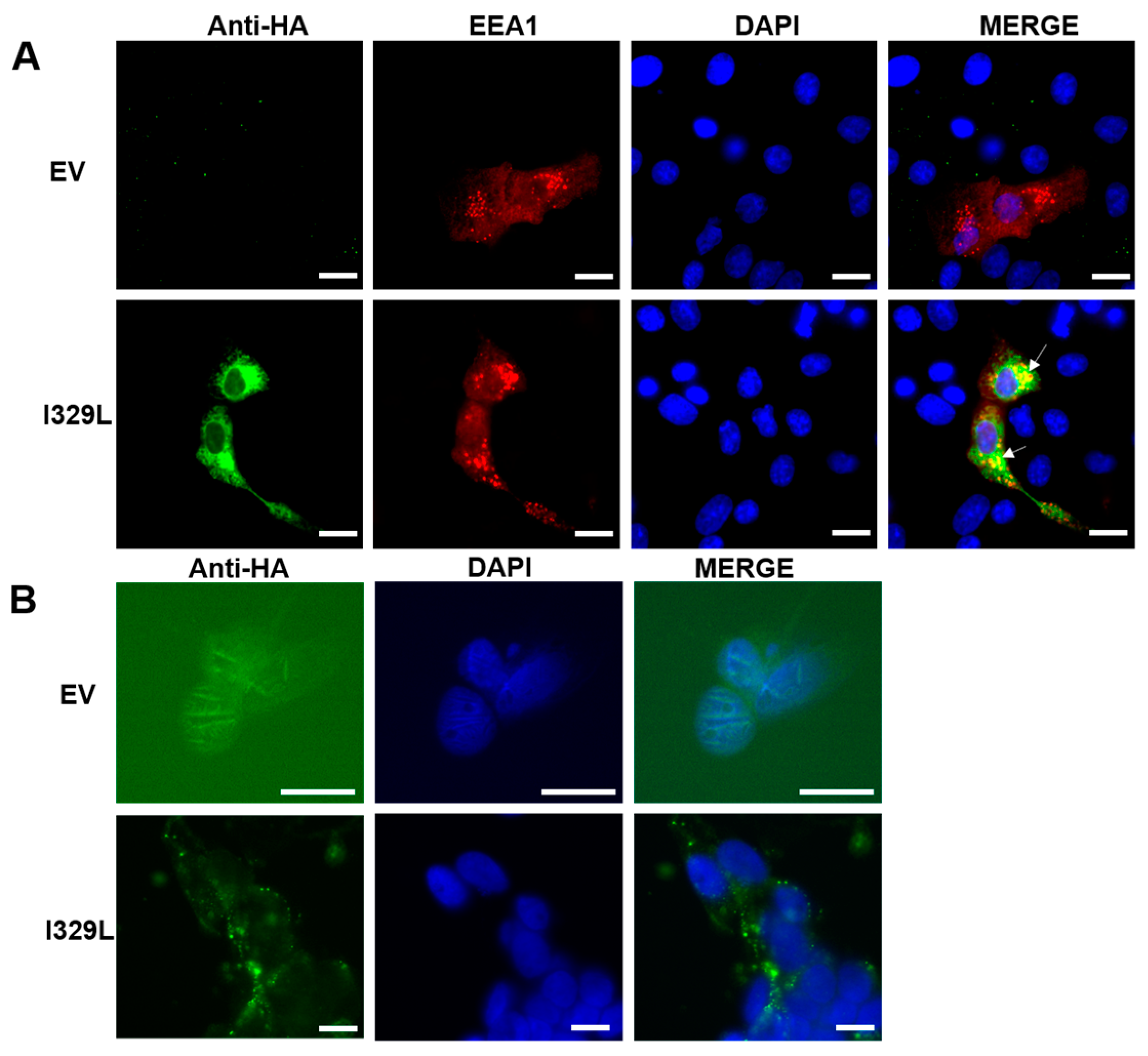
3.1. I329L Localizes to the Cell Membrane and Early Endosomes
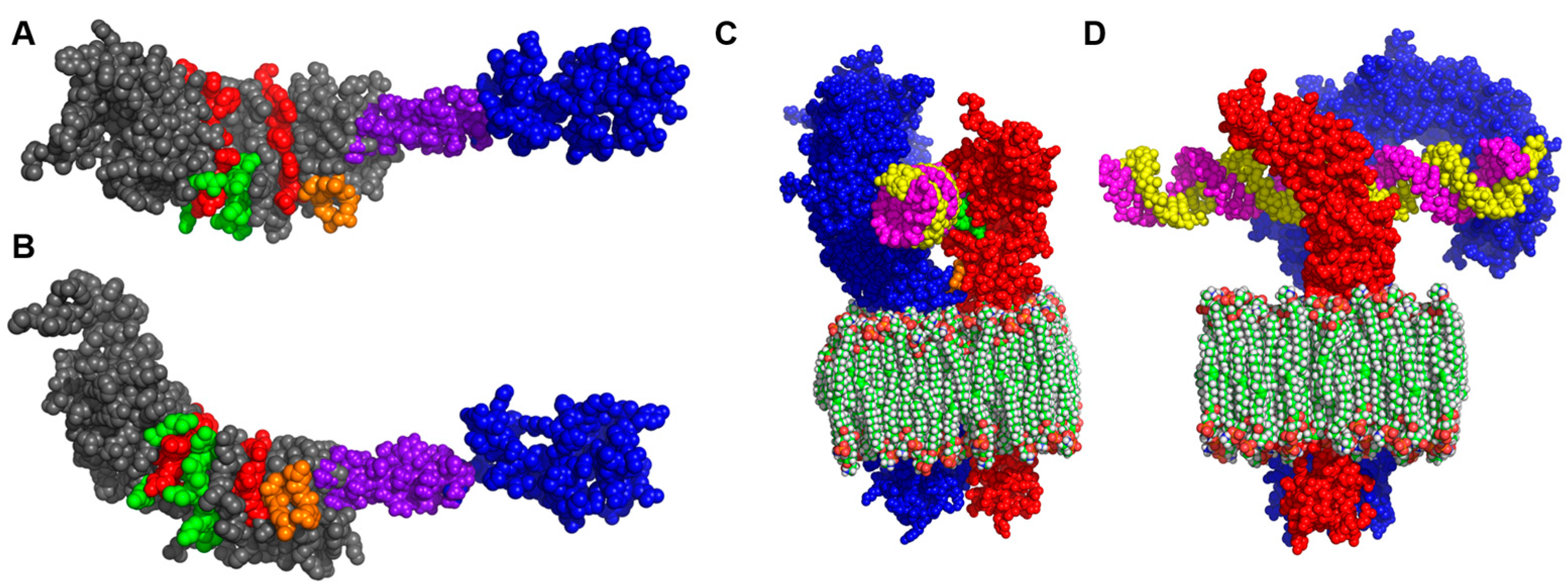
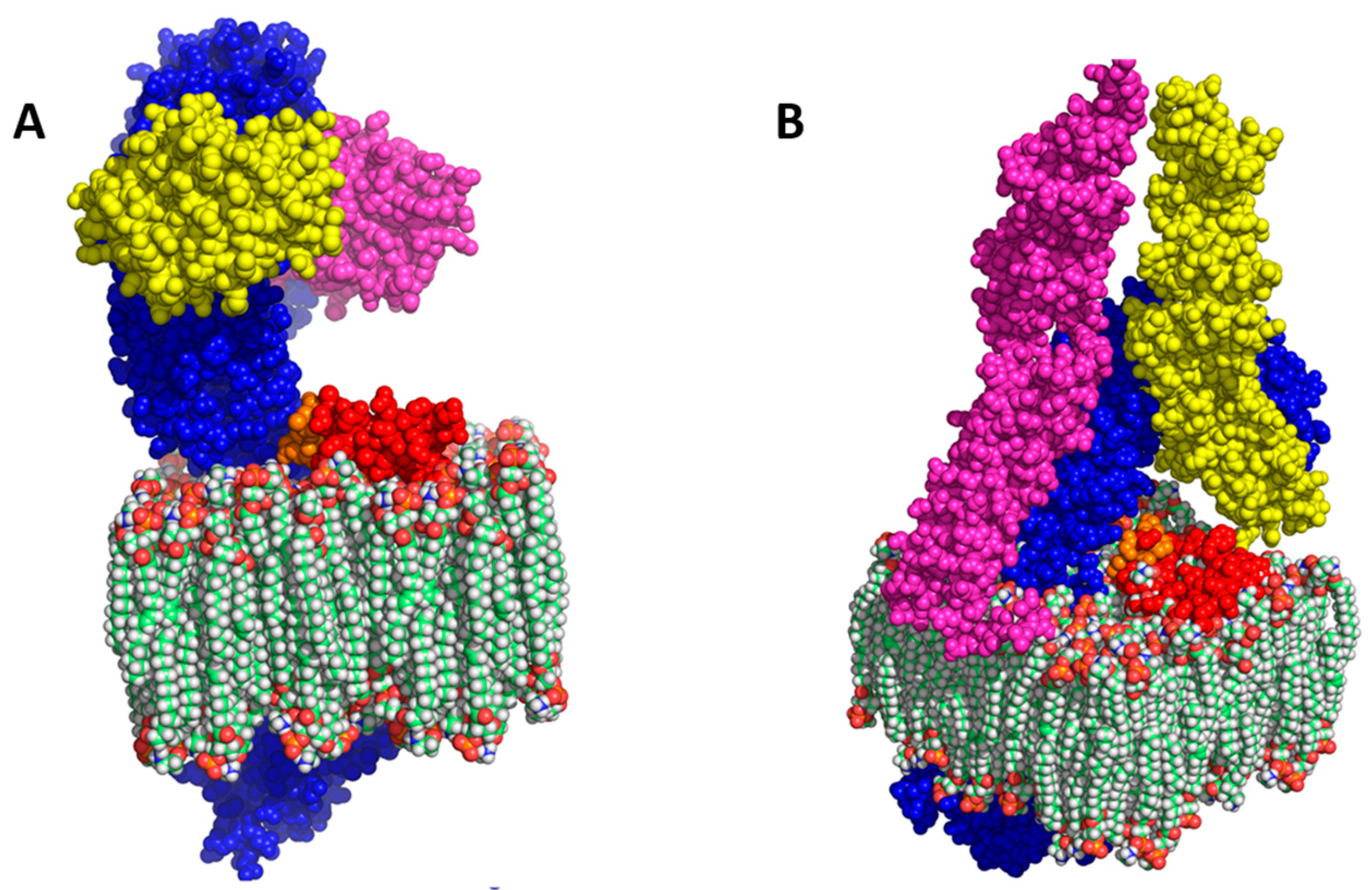
3.2. Homology Modelling of I329L Reveals Distinct Functional Domains
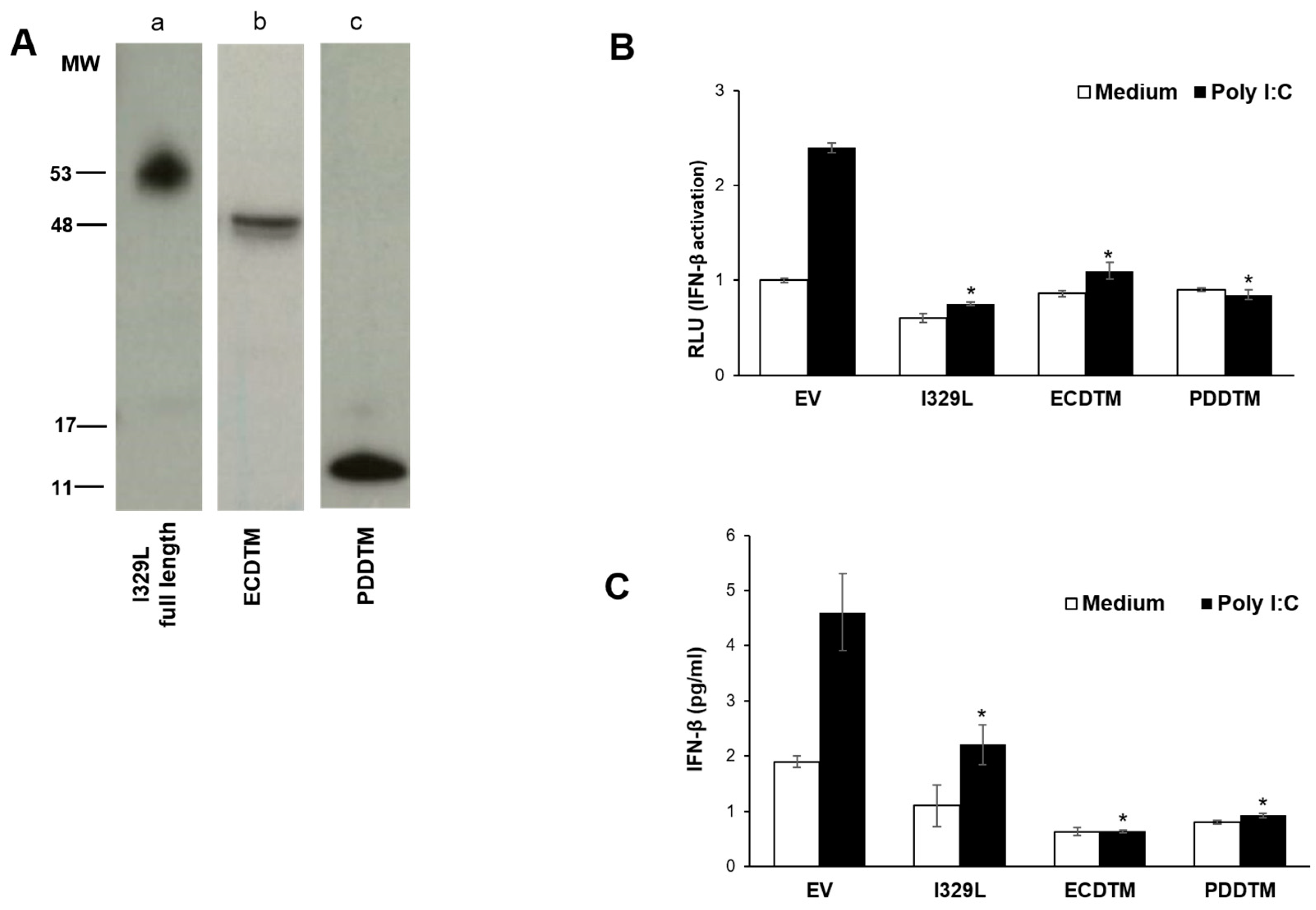
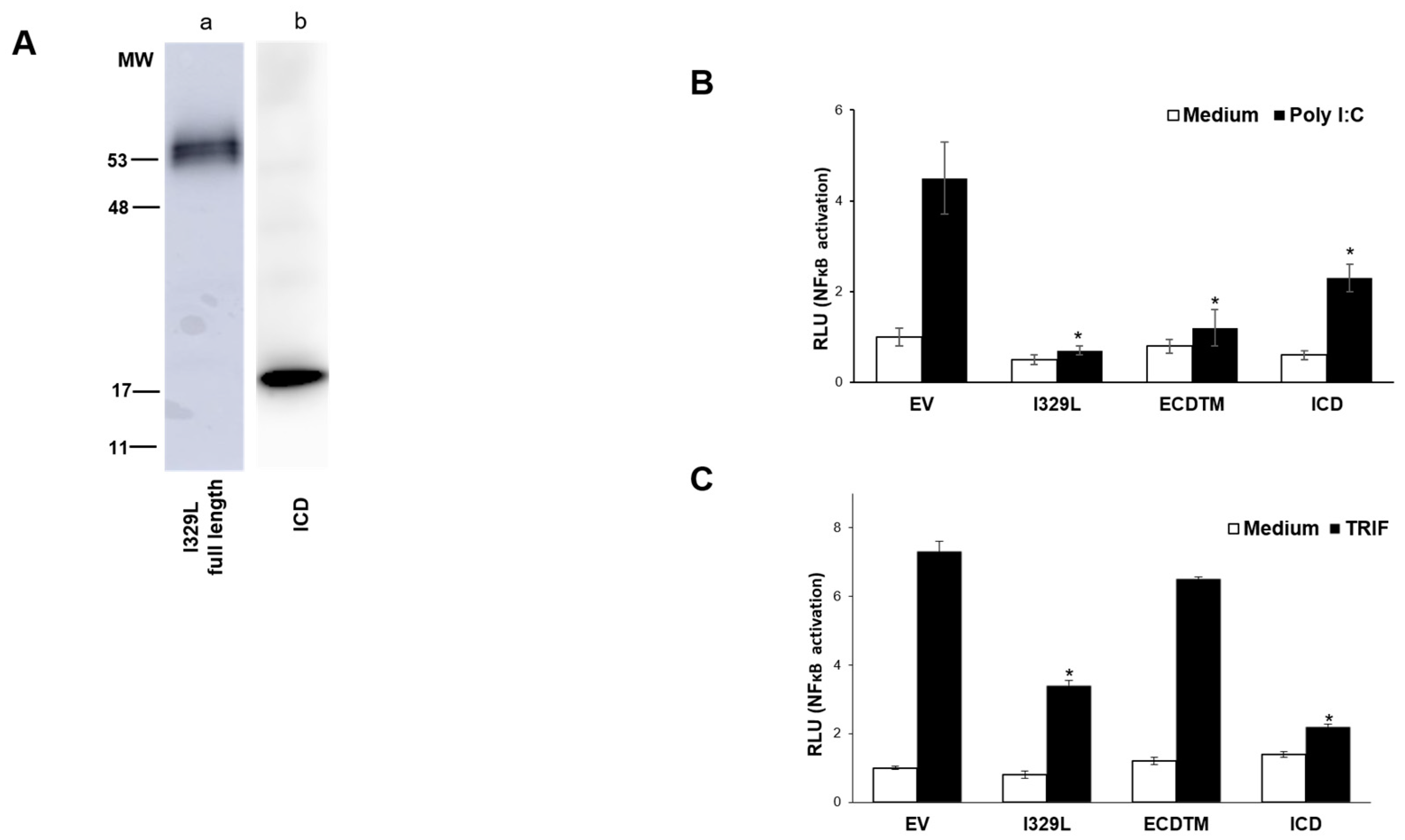
3.3. The I329L Extracellular Domain Interferes with TLR3 Signalling through a Putative Dimerization Domain (PDD)
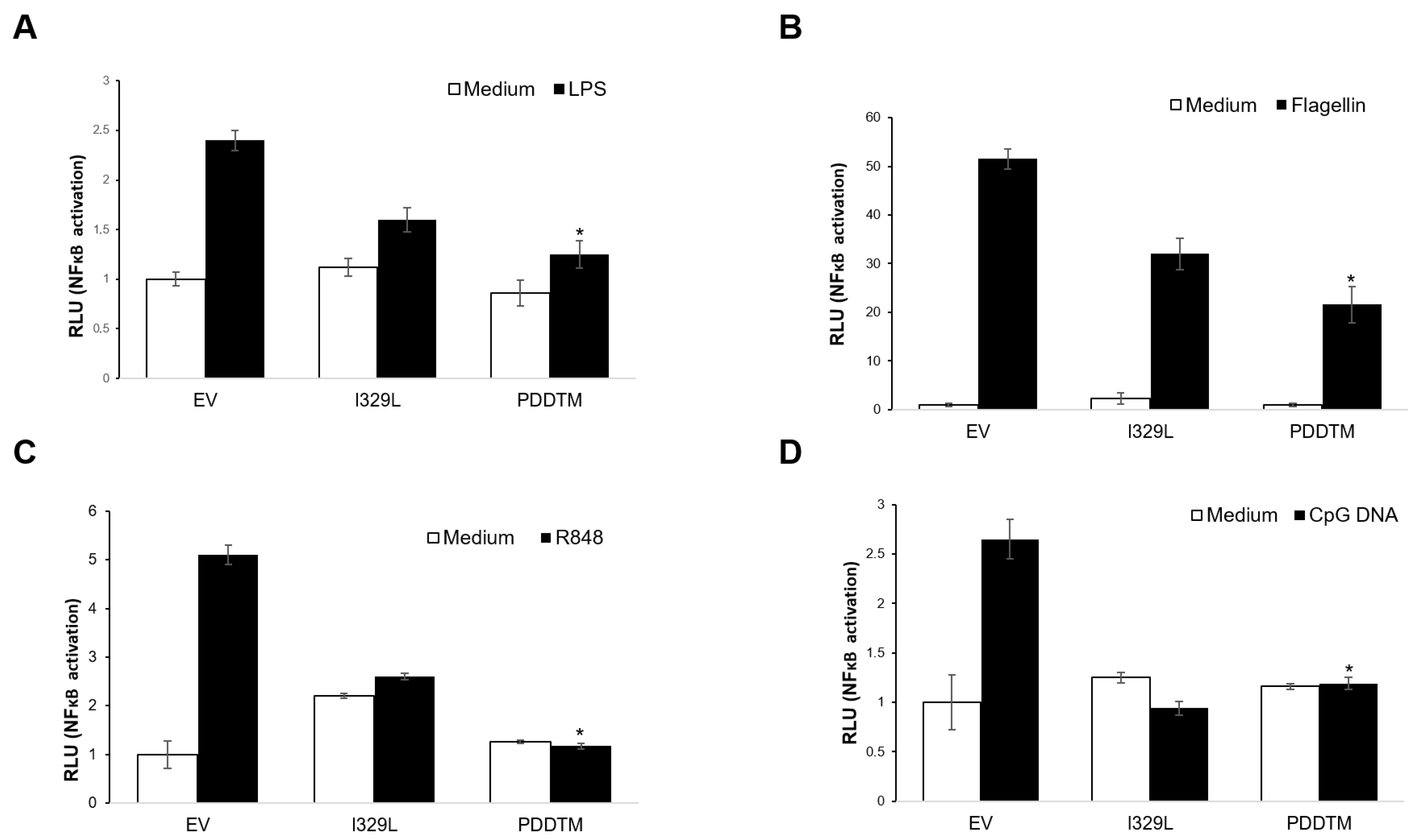
3.4. I329L Functions as a General Inhibitor of TLR Signalling via Its PDDTM
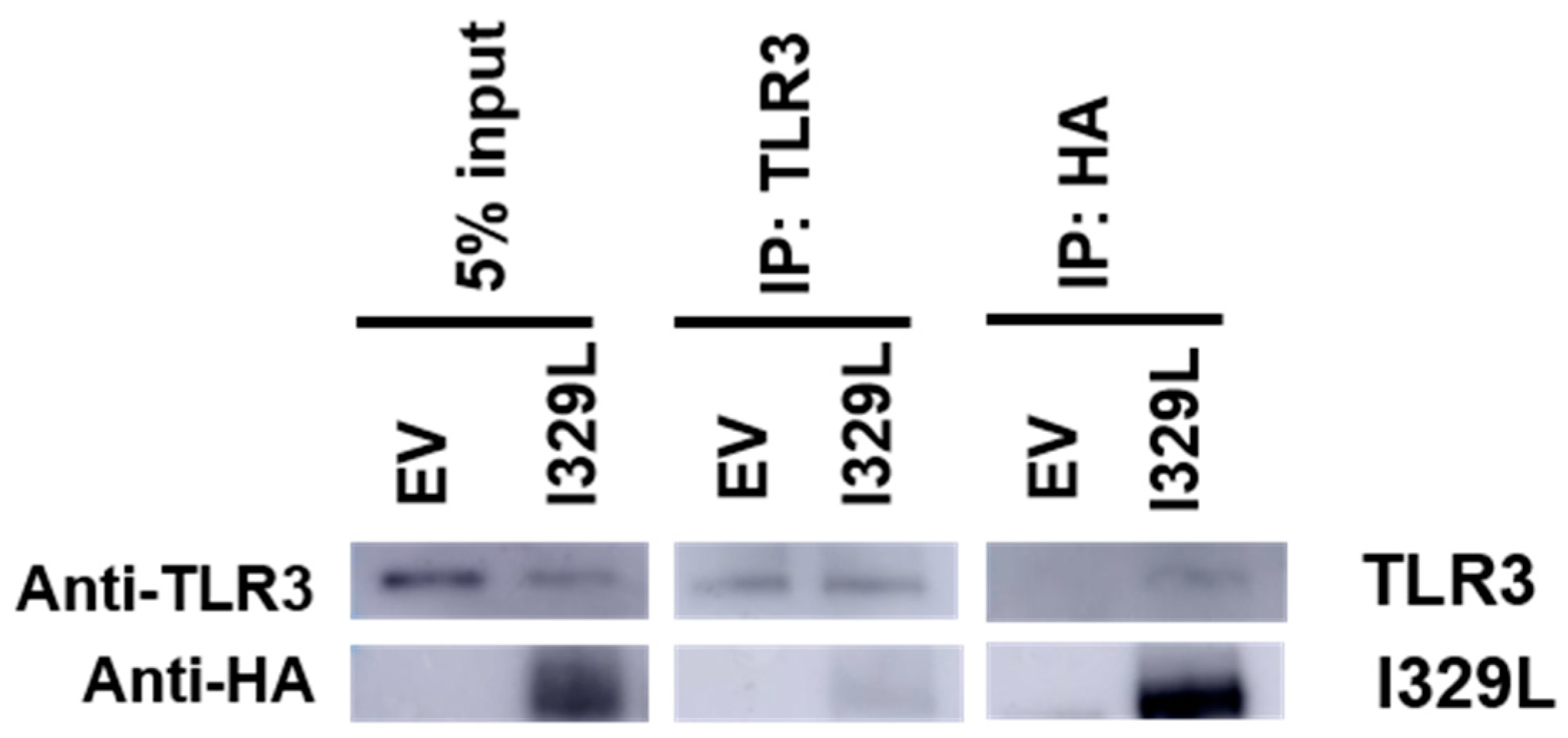
3.5. Association of I329L and TLR3
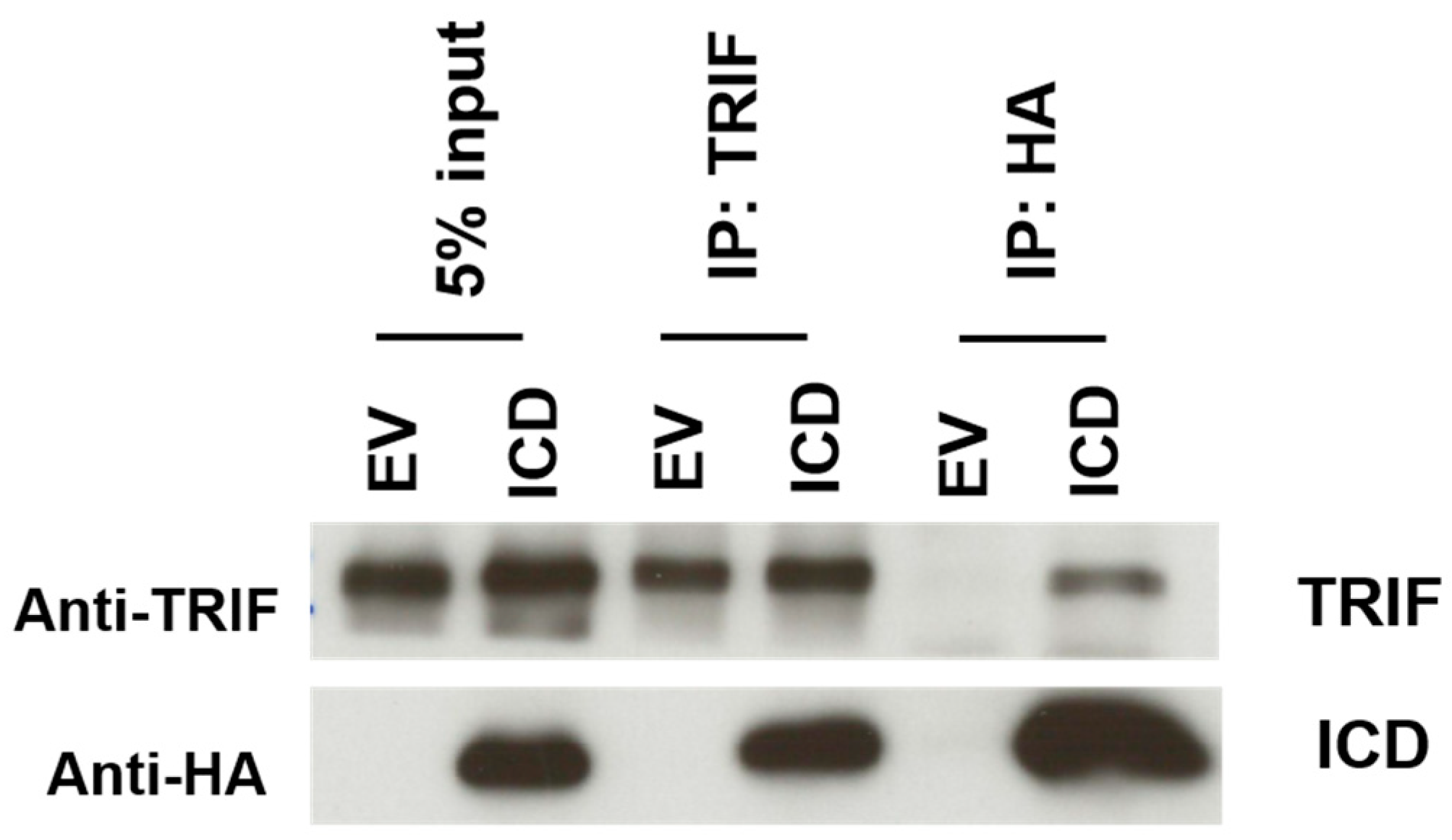
3.6. The I329L Protein Inhibits TLR3 and TLR4 Signalling through Direct Interaction with TRIF
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tulman, E.R.; Delhon, G.A.; Ku, B.K.; Rock, D.L. African Swine Fever Virus. In Lesser Known Large dsDNA Viruses; Current Topics in Microbiology and Immunology; Van Etten, J.L., Ed.; Springer: Berlin/Heidelberg, Germany, 2009; pp. 43–87. ISBN 978-3-540-68618-7. [Google Scholar]
- Penrith, M.L.; Kivaria, F.M. One Hundred Years of African Swine Fever in Africa: Where Have We Been, Where Are We Now, Where Are We Going? Transbound. Emerg. Dis. 2022, 69, e1179–e1200. [Google Scholar] [CrossRef] [PubMed]
- Correia, S.; Ventura, S.; Parkhouse, R.M. Identification and Utility of Innate Immune System Evasion Mechanisms of ASFV. Virus Res. 2013, 173, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.J. African Swine Fever Vaccinology: The Biological Challenges from Immunological Perspectives. Viruses 2022, 14, 2021. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Hopwood, P.; Abrams, C.C.; Downing, A.; Murray, F.; Talbot, R.; Archibald, A.; Lowden, S.; Dixon, L.K. Macrophage Transcriptional Responses Following In Vitro Infection with a Highly Virulent African Swine Fever Virus Isolate. J. Virol. 2006, 80, 10514–10521. [Google Scholar] [CrossRef] [PubMed]
- He, W.-R.; Yuan, J.; Ma, Y.-H.; Zhao, C.-Y.; Yang, Z.-Y.; Zhang, Y.; Han, S.; Wan, B.; Zhang, G.-P. Modulation of Host Antiviral Innate Immunity by African Swine Fever Virus: A Review. Animals 2022, 12, 2935. [Google Scholar] [CrossRef]
- de Oliveira, V.L.; Almeida, S.C.P.; Soares, H.R.; Crespo, A.; Marshall-Clarke, S.; Parkhouse, R.M.E. A Novel TLR3 Inhibitor Encoded by African Swine Fever Virus (ASFV). Arch. Virol. 2011, 156, 597–609. [Google Scholar] [CrossRef]
- Reis, A.L.; Goatley, L.C.; Jabbar, T.; Lopez, E.; Rathakrishnan, A.; Dixon, L.K. Deletion of the Gene for the Type I Interferon Inhibitor I329L from the Attenuated African Swine Fever Virus OURT88/3 Strain Reduces Protection Induced in Pigs. Vaccines 2020, 8, 262. [Google Scholar] [CrossRef]
- Henriques, E.S.; Brito, R.M.M.; Soares, H.; Ventura, S.; de Oliveira, V.L.; Parkhouse, R.M.E. Modeling of the Toll-like Receptor 3 and a Putative Toll-like Receptor 3 Antagonist Encoded by the African Swine Fever Virus. Protein Sci. 2011, 20, 247–255. [Google Scholar] [CrossRef]
- Kelley, L.A.; Sternberg, M.J.E. Protein Structure Prediction on the Web: A Case Study Using the Phyre Server. Nat. Protoc. 2009, 4, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. I-TASSER Server for Protein 3D Structure Prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef]
- Eswar, N.; Webb, B.; Marti-Renom, M.A.; Madhusudhan, M.S.; Eramian, D.; Shen, M.; Pieper, U.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Protein Sci. 2007, 50, 2.9.1–2.9.31. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A Program to Check the Stereochemical Quality of Protein Structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Ikeda, Y.; Collins, M.K.L.; Radcliffe, P.A.; Mitrophanous, K.A.; Takeuchi, Y. Gene Transduction Efficiency in Cells of Different Species by HIV and EIAV Vectors. Gene 2002, 9, 932–938. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Botos, I.; Wang, Y.; Leonard, J.N.; Shiloach, J.; Segal, D.M.; Davies, D.R. Structural Basis of Toll-Like Receptor 3 Signaling with Double-Stranded RNA. Science 2008, 320, 379–381. [Google Scholar] [CrossRef] [PubMed]
- Pirher, N.; Ivičak, K.; Pohar, J.; Benčina, M.; Jerala, R. A Second Binding Site for Double-Stranded RNA in TLR3 and Consequences for Interferon Activation. Nat. Struct. Mol. Biol. 2008, 15, 761–763. [Google Scholar] [CrossRef] [PubMed]
- Parkhouse, R.M. Three B-Cell Surface Molecules Associating with Membrane Immunoglobulin. Immunology 1990, 69, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, Z.; Ge, S.; Zuo, Y.; Lu, H.; Lv, Y.; Han, N.; Cai, Y.; Wu, X.; Wang, Z. Attenuated African Swine Fever Virus through Serial Passaging of Viruses in Cell Culture: A Brief Review on the Knowledge Gathered during 60 Years of Research. Virus Genes 2022, 59, 13–24. [Google Scholar] [CrossRef]
- Gallardo, C.; Sánchez, E.G.; Pérez-Núñez, D.; Nogal, M.; de León, P.; Carrascosa, Á.L.; Nieto, R.; Soler, A.; Arias, M.L.; Revilla, Y. African Swine Fever Virus (ASFV) Protection Mediated by NH/P68 and NH/P68 Recombinant Live-Attenuated Viruses. Vaccine 2018, 36, 2694–2704. [Google Scholar] [CrossRef] [PubMed]
- Gladue, D.P.; O’Donnell, V.; Ramirez-Medina, E.; Rai, A.; Pruitt, S.; Vuono, E.A.; Silva, E.; Velazquez-Salinas, L.; Borca, M.V. Deletion of CD2-Like (CD2v) and C-Type Lectin-Like (EP153R) Genes from African Swine Fever Virus Georgia-∆9GL Abrogates Its Effectiveness as an Experimental Vaccine. Viruses 2020, 12, 1185. [Google Scholar] [CrossRef]
- Kondo, T.; Kawai, T.; Akira, S. Dissecting Negative Regulation of Toll-like Receptor Signaling. Trends Immunol. 2012, 33, 449–458. [Google Scholar] [CrossRef]
- Anderson, J.; Olafsdottir, T.A.; Kratochvil, S.; McKay, P.F.; Östensson, M.; Persson, J.; Shattock, R.J.; Harandi, A.M. Molecular Signatures of a TLR4 Agonist-Adjuvanted HIV-1 Vaccine Candidate in Humans. Front. Immunol. 2018, 9, 301. [Google Scholar] [CrossRef] [PubMed]
- Connolly, D.J.; O’Neill, L.A. New Developments in Toll-like Receptor Targeted Therapeutics. Curr. Opin. Pharmacol. 2012, 12, 510–518. [Google Scholar] [CrossRef]
- Szabo, A.; Gogolak, P.; Pazmandi, K.; Kis-Toth, K.; Riedl, K.; Wizel, B.; Lingnau, K.; Bacsi, A.; Rethi, B.; Rajnavolgyi, E. The Two-Component Adjuvant IC31® Boosts Type I Interferon Production of Human Monocyte-Derived Dendritic Cells via Ligation of Endosomal TLRs. PLoS ONE 2013, 8, e55264. [Google Scholar] [CrossRef]
- Bunting, R.A.; Duffy, K.E.; Lamb, R.J.; San Mateo, L.R.; Smalley, K.; Raymond, H.; Liu, X.; Petley, T.; Fisher, J.; Beck, H.; et al. Novel Antagonist Antibody to TLR3 Blocks Poly(I:C)-Induced Inflammation in Vivo and in Vitro. Cell. Immunol. 2011, 267, 9–16. [Google Scholar] [CrossRef]
- Samuel, C.E. Antiviral Actions of Interferons. Clin. Microbiol. Rev. 2001, 14, 778–809. [Google Scholar] [CrossRef]
- Randall, R.E.; Goodbourn, S. Interferons and Viruses: An Interplay between Induction, Signalling, Antiviral Responses and Virus Countermeasures. J. Gen. Virol. 2008, 89, 1–47. [Google Scholar] [CrossRef] [PubMed]
- Afonso, C.L.; Piccone, M.E.; Zaffuto, K.M.; Neilan, J.; Kutish, G.F.; Lu, Z.; Balinsky, C.A.; Gibb, T.R.; Bean, T.J.; Zsak, L.; et al. African Swine Fever Virus Multigene Family 360 and 530 Genes Affect Host Interferon Response. J. Virol. 2004, 78, 1858–1864. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Xue, Y.; Niu, T.; Li, X.; Cheng, M.; Bao, M.; Zou, B.; Shi, C.; Wang, J.; Yang, W.; et al. African Swine Fever Virus MGF505-7R Protein Interacted with IRF7and TBK1 to Inhibit Type I Interferon Production. Virus Res. 2022, 322, 198931. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Correia, S.; Moura, P.L.; Ventura, S.; Leitão, A.; Parkhouse, R.M.E. I329L: A Dual Action Viral Antagonist of TLR Activation Encoded by the African Swine Fever Virus (ASFV). Viruses 2023, 15, 445. https://doi.org/10.3390/v15020445
Correia S, Moura PL, Ventura S, Leitão A, Parkhouse RME. I329L: A Dual Action Viral Antagonist of TLR Activation Encoded by the African Swine Fever Virus (ASFV). Viruses. 2023; 15(2):445. https://doi.org/10.3390/v15020445
Chicago/Turabian StyleCorreia, Sílvia, Pedro Luís Moura, Sónia Ventura, Alexandre Leitão, and Robert Michael Evans Parkhouse. 2023. "I329L: A Dual Action Viral Antagonist of TLR Activation Encoded by the African Swine Fever Virus (ASFV)" Viruses 15, no. 2: 445. https://doi.org/10.3390/v15020445
APA StyleCorreia, S., Moura, P. L., Ventura, S., Leitão, A., & Parkhouse, R. M. E. (2023). I329L: A Dual Action Viral Antagonist of TLR Activation Encoded by the African Swine Fever Virus (ASFV). Viruses, 15(2), 445. https://doi.org/10.3390/v15020445