

Enhanced Viral Metagenomics with Lazypipe 2

 , ,
, ,  ,
,  and
and
Abstract
1. Introduction
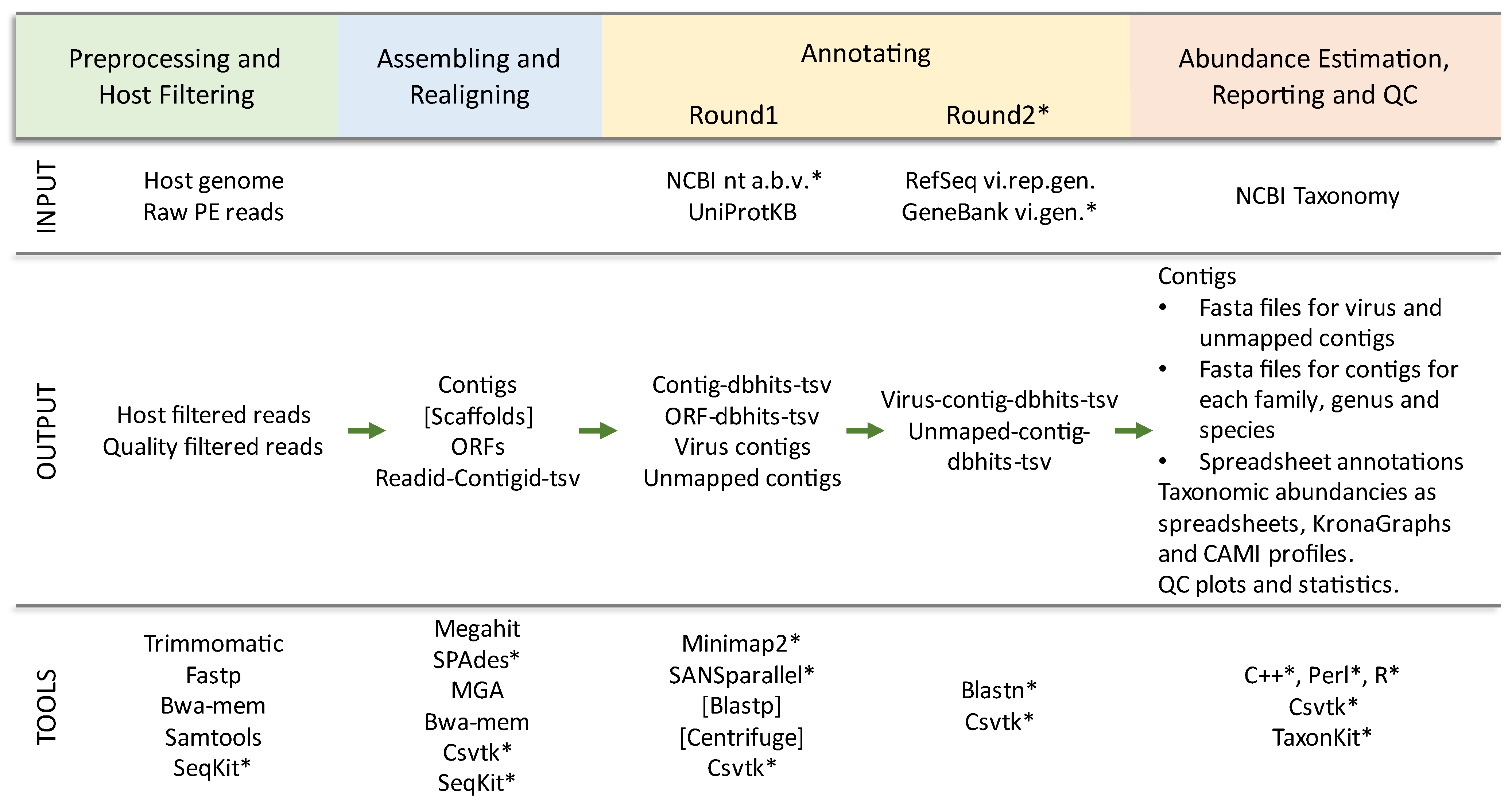
2. Materials and Methods
2.1. Code Updates
2.1.1. Code Restructuring for Better Stability and Transparency
2.1.2. Support for Parallel Analysis of Large Data Collections
2.2. New Features
2.2.1. Integrating SPAdes
2.2.2. Integrating Minimap2 Aligner
2.2.3. Integrating Blastn for Second Round Annotations
2.2.4. Improved Bacteriophage Labelling and Annotation
2.2.5. New Interface with Snakemake
2.3. Benchmarking
2.3.1. Human Simulated Metagenome
2.3.2. Viral Genomes Associated with the Domestic Dog
2.3.3. Bacterial Genomes Associated with the Domestic Dog
2.3.4. Canine Simulated Metagenome
3. Results
3.1. Benchmarking Taxonomic Profiling
3.1.1. Results for Human Simulated Metagenome
3.1.2. Classification Errors for Human Simulated Metagenome
3.1.3. Results for Canine Simulated Metagenome
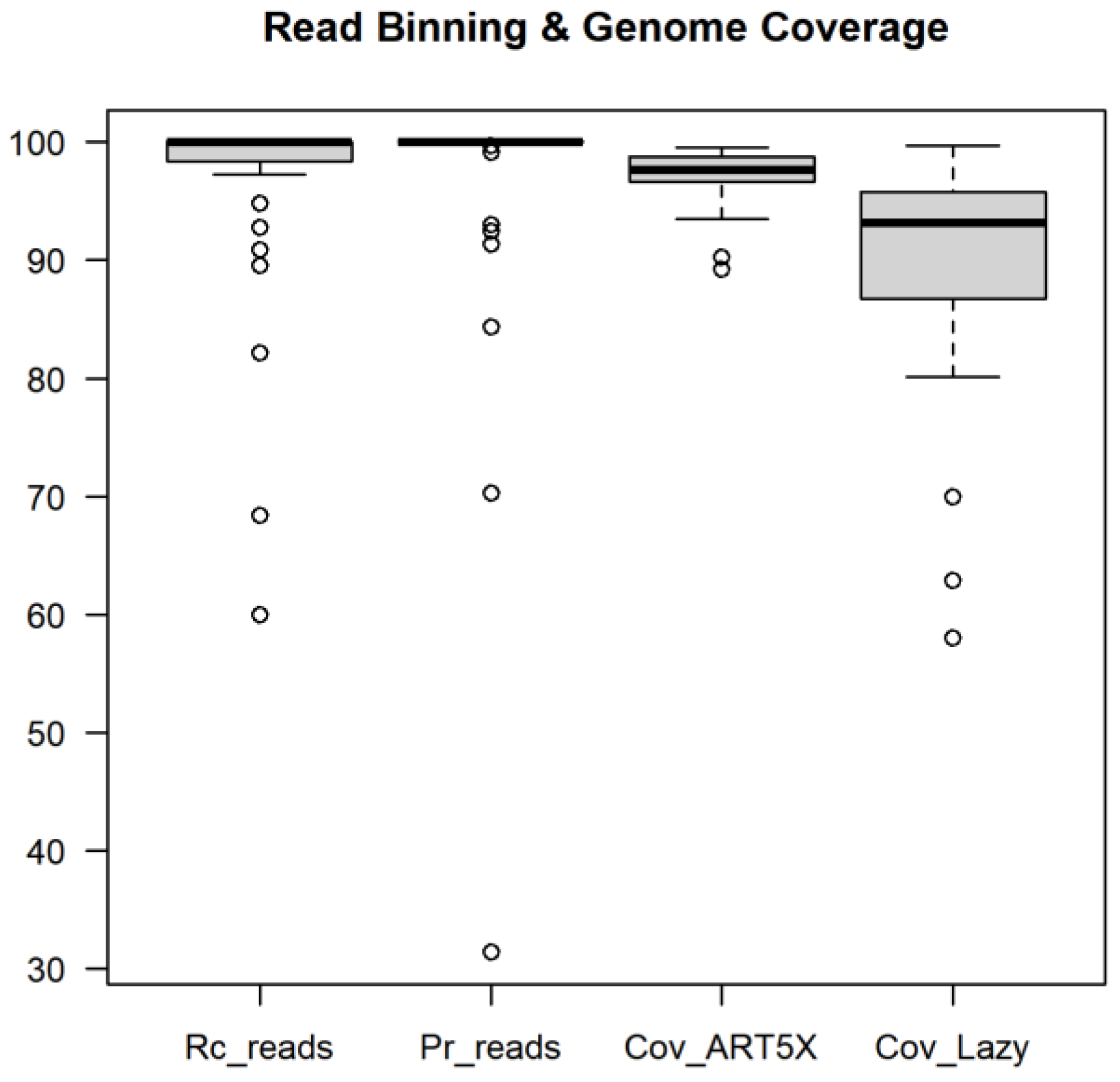
3.2. Benchmarking Read Binning and Genome Coverage
3.3. Benchmarking Time Performance and Disk Footprint
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mokili, J.L.; Rohwer, F.; Dutilh, B.E. Metagenomics and Future Perspectives in Virus Discovery. Curr. Opin. Virol. 2012, 2, 63–77. [Google Scholar] [CrossRef]
- Kalantar, K.L.; Carvalho, T.; de Bourcy, C.F.A.; Dimitrov, B.; Dingle, G.; Egger, R.; Han, J.; Holmes, O.B.; Juan, Y.-F.; King, R.; et al. IDseq-An Open Source Cloud-Based Pipeline and Analysis Service for Metagenomic Pathogen Detection and Monitoring. Gigascience 2020, 9, giaa111. [Google Scholar] [CrossRef]
- Wagner, D.D.; Marine, R.L.; Ramos, E.; Ng, T.F.F.; Castro, C.J.; Okomo-Adhiambo, M.; Harvey, K.; Doho, G.; Kelly, R.; Jain, Y.; et al. VPipe: An Automated Bioinformatics Platform for Assembly and Management of Viral Next-Generation Sequencing Data. Microbiol. Spectr. 2022, 10, e0256421. [Google Scholar] [CrossRef]
- Smits, S.L.; Bodewes, R.; Ruiz-González, A.; Baumgärtner, W.; Koopmans, M.P.; Osterhaus, A.D.M.E.; Schürch, A.C. Recovering Full-Length Viral Genomes from Metagenomes. Front. Microbiol. 2015, 6, 1069. [Google Scholar] [CrossRef]
- Graf, E.H.; Simmon, K.E.; Tardif, K.D.; Hymas, W.; Flygare, S.; Eilbeck, K.; Yandell, M.; Schlaberg, R. Unbiased Detection of Respiratory Viruses by Use of RNA Sequencing-Based Metagenomics: A Systematic Comparison to a Commercial PCR Panel. J. Clin. Microbiol. 2016, 54, 1000–1007. [Google Scholar] [CrossRef]
- Pallen, M.J. Diagnostic Metagenomics: Potential Applications to Bacterial, Viral and Parasitic Infections. Parasitology 2014, 141, 1856–1862. [Google Scholar] [CrossRef] [PubMed]
- de Vries, J.J.C.; Brown, J.R.; Fischer, N.; Sidorov, I.A.; Morfopoulou, S.; Huang, J.; Munnink, B.B.O.; Sayiner, A.; Bulgurcu, A.; Rodriguez, C.; et al. Benchmark of Thirteen Bioinformatic Pipelines for Metagenomic Virus Diagnostics Using Datasets from Clinical Samples. J. Clin. Virol. 2021, 141, 104908. [Google Scholar] [CrossRef]
- Lorenzi, H.A.; Hoover, J.; Inman, J.; Safford, T.; Murphy, S.; Kagan, L.; Williamson, S.J. TheViral MetaGenome Annotation Pipeline(VMGAP):An Automated Tool for the Functional Annotation of Viral Metagenomic Shotgun Sequencing Data. Stand. Genom. Sci. 2011, 4, 418–429. [Google Scholar] [CrossRef]
- Kostic, A.D.; Ojesina, A.I.; Pedamallu, C.S.; Jung, J.; Verhaak, R.G.W.; Getz, G.; Meyerson, M. PathSeq: Software to Identify or Discover Microbes by Deep Sequencing of Human Tissue. Nat. Biotechnol. 2011, 29, 393–396. [Google Scholar] [CrossRef]
- Wommack, K.E.; Bhavsar, J.; Polson, S.W.; Chen, J.; Dumas, M.; Srinivasiah, S.; Furman, M.; Jamindar, S.; Nasko, D.J. VIROME: A Standard Operating Procedure for Analysis of Viral Metagenome Sequences. Stand. Genom. Sci. 2012, 6, 427–439. [Google Scholar] [CrossRef]
- Naeem, R.; Rashid, M.; Pain, A. READSCAN: A Fast and Scalable Pathogen Discovery Program with Accurate Genome Relative Abundance Estimation. Bioinformatics 2013, 29, 391–392. [Google Scholar] [CrossRef]
- Wang, Q.; Jia, P.; Zhao, Z. VirusFinder: Software for Efficient and Accurate Detection of Viruses and Their Integration Sites in Host Genomes through next Generation Sequencing Data. PLoS ONE 2013, 8, e64465. [Google Scholar] [CrossRef]
- Naccache, S.N.; Federman, S.; Veeraraghavan, N.; Zaharia, M.; Lee, D.; Samayoa, E.; Bouquet, J.; Greninger, A.L.; Luk, K.-C.; Enge, B.; et al. A Cloud-Compatible Bioinformatics Pipeline for Ultrarapid Pathogen Identification from next-Generation Sequencing of Clinical Samples. Genome Res. 2014, 24, 1180–1192. [Google Scholar] [CrossRef]
- Roux, S.; Tournayre, J.; Mahul, A.; Debroas, D.; Enault, F. Metavir 2: New Tools for Viral Metagenome Comparison and Assembled Virome Analysis. BMC Bioinform. 2014, 15, 76. [Google Scholar] [CrossRef]
- Li, Y.; Wang, H.; Nie, K.; Zhang, C.; Zhang, Y.; Wang, J.; Niu, P.; Ma, X. VIP: An Integrated Pipeline for Metagenomics of Virus Identification and Discovery. Sci. Rep. 2016, 6, 23774. [Google Scholar] [CrossRef]
- Fosso, B.; Santamaria, M.; D’Antonio, M.; Lovero, D.; Corrado, G.; Vizza, E.; Passaro, N.; Garbuglia, A.; Capobianchi, M.; Crescenzi, M.; et al. MetaShot: An Accurate Workflow for Taxon Classification of Host-Associated Microbiome from Shotgun Metagenomic Data. Bioinformatics 2017, 33, 1730–1732. [Google Scholar] [CrossRef]
- Zhao, G.; Wu, G.; Lim, E.S.; Droit, L.; Krishnamurthy, S.; Barouch, D.H.; Virgin, H.W.; Wang, D. VirusSeeker, a Computational Pipeline for Virus Discovery and Virome Composition Analysis. Virology 2017, 503, 21–30. [Google Scholar] [CrossRef]
- Bhuvaneshwar, K.; Song, L.; Madhavan, S.; Gusev, Y. ViGEN: An Open Source Pipeline for the Detection and Quantification of Viral RNA in Human Tumors. Front. Microbiol. 2018, 9, 1172. [Google Scholar] [CrossRef]
- Vilsker, M.; Moosa, Y.; Nooij, S.; Fonseca, V.; Ghysens, Y.; Dumon, K.; Pauwels, R.; Alcantara, L.C.; Vanden Eynden, E.; Vandamme, A.-M.; et al. Genome Detective: An Automated System for Virus Identification from High-Throughput Sequencing Data. Bioinformatics 2019, 35, 871–873. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved Metagenomic Analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef]
- Pérot, P.; Bigot, T.; Temmam, S.; Regnault, B.; Eloit, M. Microseek: A Protein-Based Metagenomic Pipeline for Virus Diagnostic and Discovery. Viruses 2022, 14, 1990. [Google Scholar] [CrossRef] [PubMed]
- Plyusnin, I.; Kant, R.; Jääskeläinen, A.J.; Sironen, T.; Holm, L.; Vapalahti, O.; Smura, T. Novel NGS Pipeline for Virus Discovery from a Wide Spectrum of Hosts and Sample Types. Virus Evol. 2020, 6, veaa091. [Google Scholar] [CrossRef] [PubMed]
- Kuivanen, S.; Levanov, L.; Kareinen, L.; Sironen, T.; Jääskeläinen, A.J.; Plyusnin, I.; Zakham, F.; Emmerich, P.; Schmidt-Chanasit, J.; Hepojoki, J.; et al. Detection of Novel Tick-Borne Pathogen, Alongshan Virus, in Ixodes Ricinus Ticks, South-Eastern Finland, 2019. Eurosurveillance 2019, 24, 1900394. [Google Scholar] [CrossRef] [PubMed]
- Zakham, F.; Albalawi, A.E.; Alanazi, A.D.; Truong Nguyen, P.; Alouffi, A.S.; Alaoui, A.; Sironen, T.; Smura, T.; Vapalahti, O. Viral RNA Metagenomics of Hyalomma Ticks Collected from Dromedary Camels in Makkah Province, Saudi Arabia. Viruses 2021, 13, 1396. [Google Scholar] [CrossRef]
- Truong Nguyen, P.T.; Culverwell, C.L.; Suvanto, M.T.; Korhonen, E.M.; Uusitalo, R.; Vapalahti, O.; Smura, T.; Huhtamo, E. Characterisation of the RNA Virome of Nine Ochlerotatus Species in Finland. Viruses 2022, 14, 1489. [Google Scholar] [CrossRef]
- Virtanen, J.; Zalewski, A.; Kołodziej-Sobocińska, M.; Brzeziński, M.; Smura, T.; Sironen, T. Diversity and Transmission of Aleutian Mink Disease Virus in Feral and Farmed American Mink and Native Mustelids. Virus Evol. 2021, 7, veab075. [Google Scholar] [CrossRef]
- Forbes, K.M.; Webala, P.W.; Jääskeläinen, A.J.; Abdurahman, S.; Ogola, J.; Masika, M.M.; Kivistö, I.; Alburkat, H.; Plyusnin, I.; Levanov, L.; et al. Bombali Virus in Mops Condylurus Bat, Kenya. Emerg. Infect. Dis. 2019, 25, 955–957. [Google Scholar] [CrossRef]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes De Novo Assembler. Curr. Protoc. Bioinform. 2020, 70, e102. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- BLAST: Basic Local Alignment Search Tool. Available online: https://blast.ncbi.nlm.nih.gov/ (accessed on 31 October 2022).
- Mölder, F.; Jablonski, K.P.; Letcher, B.; Hall, M.B.; Tomkins-Tinch, C.H.; Sochat, V.; Forster, J.; Lee, S.; Twardziok, S.O.; Kanitz, A.; et al. Sustainable Data Analysis with Snakemake. F1000Research 2021, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Ren, H. TaxonKit: A Practical and Efficient NCBI Taxonomy Toolkit. J. Genet. Genom. 2021, 48, 844–850. [Google Scholar] [CrossRef] [PubMed]
- Csvtk—CSV/TSV Toolkit. Available online: https://bioinf.shenwei.me/csvtk/ (accessed on 21 December 2022).
- Somervuo, P.; Holm, L. SANSparallel: Interactive Homology Search against Uniprot. Nucleic Acids Res. 2015, 43, W24–W29. [Google Scholar] [CrossRef] [PubMed]
- Törönen, P.; Holm, L. PANNZER-A Practical Tool for Protein Function Prediction. Protein Sci. 2022, 31, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Sutton, T.D.S.; Clooney, A.G.; Ryan, F.J.; Ross, R.P.; Hill, C. Choice of Assembly Software Has a Critical Impact on Virome Characterisation. Microbiome 2019, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Emerson, J.B.; Eloe-Fadrosh, E.A.; Sullivan, M.B. Benchmarking Viromics: An In Silico Evaluation of Metagenome-Enabled Estimates of Viral Community Composition and Diversity. PeerJ 2017, 5, e3817. [Google Scholar] [CrossRef] [PubMed]
- Mihara, T.; Nishimura, Y.; Shimizu, Y.; Nishiyama, H.; Yoshikawa, G.; Uehara, H.; Hingamp, P.; Goto, S.; Ogata, H. Linking Virus Genomes with Host Taxonomy. Viruses 2016, 8, 66. [Google Scholar] [CrossRef]
- Huang, W.; Li, L.; Myers, J.R.; Marth, G.T. ART: A next-Generation Sequencing Read Simulator. Bioinformatics 2011, 28, 593–594. [Google Scholar] [CrossRef] [PubMed]
- Geoghegan, J.L.; Holmes, E.C. Predicting Virus Emergence amid Evolutionary Noise. Open Biol. 2017, 7, 170189. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Metagenome | Host Reads | Bacterial Reads | Virus Reads | Bacterial Taxids | Virus Taxids |
|---|---|---|---|---|---|
| dog5X-ba.comp5X-vi5X | 93.37% | 6.60% | 0.021% | 58 | 39 |
| dog5X-ba5X-vi5X | 79.85% | 20.13% | 0.018% | 195 | 39 |
| dog5X-ba5X-vi4X | 79.85% | 20.13% | 0.014% | 195 | 39 |
| dog5X-ba5X-vi3X | 79.86% | 20.13% | 0.011% | 195 | 39 |
| dog5X-ba5X-vi2X | 79.86% | 20.13% | 0.007% | 195 | 39 |
| dog5X-ba5X-vi1X | 79.86% | 20.14% | 0.004% | 195 | 39 |
| Tool | Rank | True | TP | FP | FN | Pr | Rc | F1 |
|---|---|---|---|---|---|---|---|---|
| Lazypipe 2 --ann minimap | species | 83 | 79 | 2 | 4 | 97.5% | 95.2% | 96.3% |
| IDseq | species | 83 | 77 | 4 | 6 | 95.1% | 92.8% | 93.9% |
| GenomeDetectiveVirus * | species | 80 | 75 | 5 | 5 | 93.8% | 93.8% | 93.8% |
| Lazypipe 2 --ann sans | species | 83 | 74 | 13 | 9 | 85.1% | 89.2% | 87.1% |
| Lazypipe1 --ann sans | species | 83 | 73 | 13 | 10 | 84.9% | 88.0% | 86.4% |
| Kraken2 | species | 83 | 75 | 18 | 8 | 80.6% | 90.4% | 85.2% |
| Lazypipe 2 --ann minimap | genus | 48 | 47 | 0 | 1 | 100.0% | 97.9% | 98.9% |
| IDseq | genus | 48 | 46 | 0 | 2 | 100.0% | 95.8% | 97.9% |
| GenomeDetectiveVirus * | genus | 48 | 46 | 1 | 2 | 97.9% | 95.8% | 96.8% |
| Kraken2 | genus | 48 | 46 | 1 | 2 | 97.9% | 95.8% | 96.8% |
| Lazypipe 2 --ann sans | genus | 48 | 44 | 1 | 4 | 97.8% | 91.7% | 94.6% |
| Lazypipe1 --ann sans | genus | 48 | 42 | 1 | 6 | 97.7% | 87.5% | 92.3% |
| Tool | Rank | True | TP | FP | FN | Pr | Rc | F1 |
|---|---|---|---|---|---|---|---|---|
| Lazypipe 2 --ann sans -t20 | species | 71 | 64 | 12 | 7 | 84.2% | 90.1% | 87.1% |
| Lazypipe 2 --ann minimap | species | 71 | 46 | 29 | 25 | 61.3% | 64.8% | 63.0% |
| Lazypipe1 --ann sans | species | 71 | 68 | 79 | 3 | 46.3% | 95.8% | 62.4% |
| IDseq | species | 71 | 42 | 24 | 29 | 63.6% | 59.2% | 61.3% |
| Lazypipe 2 --ann sans | species | 71 | 70 | 109 | 1 | 39.1% | 98.6% | 56.0% |
| Kraken2 | species | 71 | 37 | 62 | 34 | 37.4% | 52.1% | 43.5% |
| Lazypipe 2 --ann sans -t20 | genus | 44 | 41 | 4 | 3 | 91..1% | 93.2% | 92.1% |
| Lazypipe1 --ann sans | genus | 44 | 44 | 9 | 0 | 83.0% | 100.0% | 90.7% |
| Lazypipe 2 --ann minimap | genus | 44 | 34 | 5 | 10 | 87.2% | 77.3% | 81.9% |
| IDseq | genus | 44 | 33 | 3 | 11 | 91.7% | 75.0% | 82.5% |
| Lazypipe 2 --ann sans | genus | 44 | 44 | 18 | 0 | 71.0% | 100.0% | 83.0% |
| Kraken2 | genus | 44 | 29 | 9 | 15 | 76.3% | 65.9% | 70.7% |
| Virus | Lazypipe --ann sans | Lazypipe --ann minimap2 | Error Cause | Pipeline Step |
|---|---|---|---|---|
| Human endogenous retrovirus | FN | FN | Reads filtered as host reads | Preprocessing |
| Human endogenous retrovirus W | FN | FN | Preprocessing | |
| Naples phlebovirus | FN | FN | Contig (4187 nt) with high (100%) identity to Toscana phlebovirus | DB search |
| Mopeia Lassa virus reassortant 29 | FN | FN | Mis-assembled contig (1551 nt) with high (99.9%) identity to Lassa virus | Assembling |
| Hepatitis B virus | FN | TP | orf prediction | orf prediction |
| Uukuniemi uukuvirus | FN | TP | orf prediction | orf prediction |
| Influenza A virus | FN | TP | orf prediction | orf prediction |
| Mason-Pfizer monkey virus | FN | TP | Low read count | Reporting |
| Toscana phlebovirus | FP | FP | Contig (4187 nt) with high (100%) identity to FP | DB search |
| Cowpox virus | FP | TN | orfs with high (100%) identity to FP | DB search |
| Borna disease virus | FP | TN | orfs (429 nt, 606 nt and 1113 nt) with high (100%) identity to FP | DB search |
| Central chimpanzee simian foamy virus | FP | TN | orfs (668 nt and 696 nt) with high (96.0–99.0%) identity to FP | DB search |
| Eastern chimpanzee simian foamy virus | FP | TN | orfs (180–759 nt) with high (98.0–100%) identity to FP | DB search |
| African green monkey simian foamy virus | FP | TN | orf (753 nt) with high (81.1%) identity to FP | DB search |
| Human bocavirus | FP | TN | orfs (441–2016 nt) with high (100%) identity to FP | DB search |
| Chimeric Tick-borne encephalitis virus/ Dengue virus 4 | FP | TN | orf (6108 nt) with high (100%) identity to FP | DB search |
| Phlebovirus SDYY104/China/2011 virus | FP | TN | orf (6258 nt) with high (100%) identity to FP | DB search |
| SFTS phlebovirus | FP | TN | orf (738 nt) with high (100%) identity to FP | DB search |
| Bocaparvovirus sp. | FP | TN | orf (1920 nt) with high (100%) identity to FP | DB search |
| Coronavirus BtRs-BetaCoV/YN2018D | FP | TN | orf (966 nt) with high (100%) identity to FP | DB search |
| Uncultured human fecal virus | TN | FP | 12 contigs (330–586 nt) with high identity to FP | DB search |
| Metagenome | Target Taxa | TRUE | TP | FP | FN | Pr | Rc | F1 |
|---|---|---|---|---|---|---|---|---|
| dog5X-ba.comp5X-vi5X | Virus species | 35 | 35 | 1 | 0 | 97.2% | 100.0% | 98.6% |
| dog5X-ba5X-vi5X | Virus species | 35 | 35 | 1 | 0 | 97.2% | 100.0% | 98.6% |
| dog5X-ba5X-vi4X | Virus species | 35 | 35 | 1 | 0 | 97.2% | 100.0% | 98.6% |
| dog5X-ba5X-vi3X | Virus species | 35 | 35 | 1 | 0 | 97.2% | 100.0% | 98.6% |
| dog5X-ba5X-vi2X | Virus species | 35 | 35 | 1 | 0 | 97.2% | 100.0% | 98.6% |
| dog5X-ba5X-vi1X | Virus species | 35 | 34 | 1 | 1 | 97.1% | 97.1% | 97.1% |
| dog5X-ba.comp5X-vi5X | Bacterial species | 53 | 40 | 9 | 13 | 81.6% | 75.5% | 78.4% |
| dog5X-ba.comp5X-vi5X | Bacterial genera | 32 | 31 | 1 | 1 | 96.9% | 96.9% | 96.9% |
| dog5X-ba5X-vi5X | Bacterial species | 159 | 96 | 82 | 63 | 53.9% | 60.4% | 57.0% |
| dog5X-ba5X-vi5X | Bacterial genera | 71 | 61 | 3 | 10 | 95.3% | 85.9% | 90.4% |
| Tool | Real | CPU |
|---|---|---|
| Kraken2 | 0:02:38 | 0:09:13 |
| Lazypipe1 --ann sans | 1:18:02 | 6:43:42 |
| Lazypipe 2 --ann sans | 1:35:34 | 6:45:42 |
| Lazypipe 2 --ann minimap | 1:47:21 | 10:37:09 |
| Genome Detective * | 2:01:49 | NA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Plyusnin, I.; Vapalahti, O.; Sironen, T.; Kant, R.; Smura, T. Enhanced Viral Metagenomics with Lazypipe 2. Viruses 2023, 15, 431. https://doi.org/10.3390/v15020431
Plyusnin I, Vapalahti O, Sironen T, Kant R, Smura T. Enhanced Viral Metagenomics with Lazypipe 2. Viruses. 2023; 15(2):431. https://doi.org/10.3390/v15020431
Chicago/Turabian StylePlyusnin, Ilya, Olli Vapalahti, Tarja Sironen, Ravi Kant, and Teemu Smura. 2023. "Enhanced Viral Metagenomics with Lazypipe 2" Viruses 15, no. 2: 431. https://doi.org/10.3390/v15020431
APA StylePlyusnin, I., Vapalahti, O., Sironen, T., Kant, R., & Smura, T. (2023). Enhanced Viral Metagenomics with Lazypipe 2. Viruses, 15(2), 431. https://doi.org/10.3390/v15020431