

Comparative Genomics of a Polyvalent Escherichia-Salmonella Phage fp01 and In Silico Analysis of Its Receptor Binding Protein and Conserved Enterobacteriaceae Phage Receptor

 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. DNA Extraction
2.2. Sequencing and Genome Assembly
2.3. Annotation and Genome Mapping
2.4. Comparative Genomics and Phylogenetic Analysis
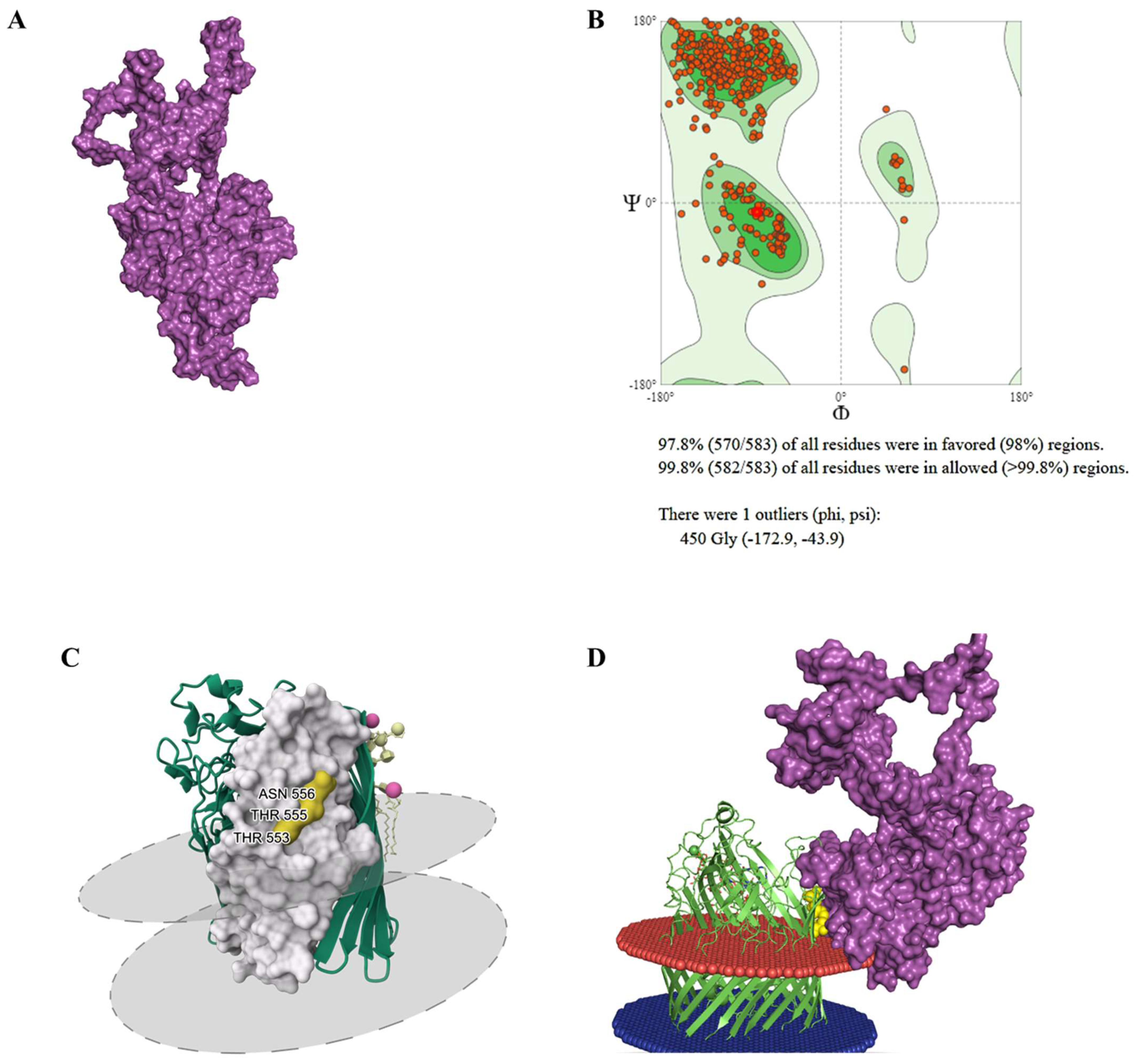
2.5. Protein Modelling and Molecular Docking
3. Results
3.1. Sequencing
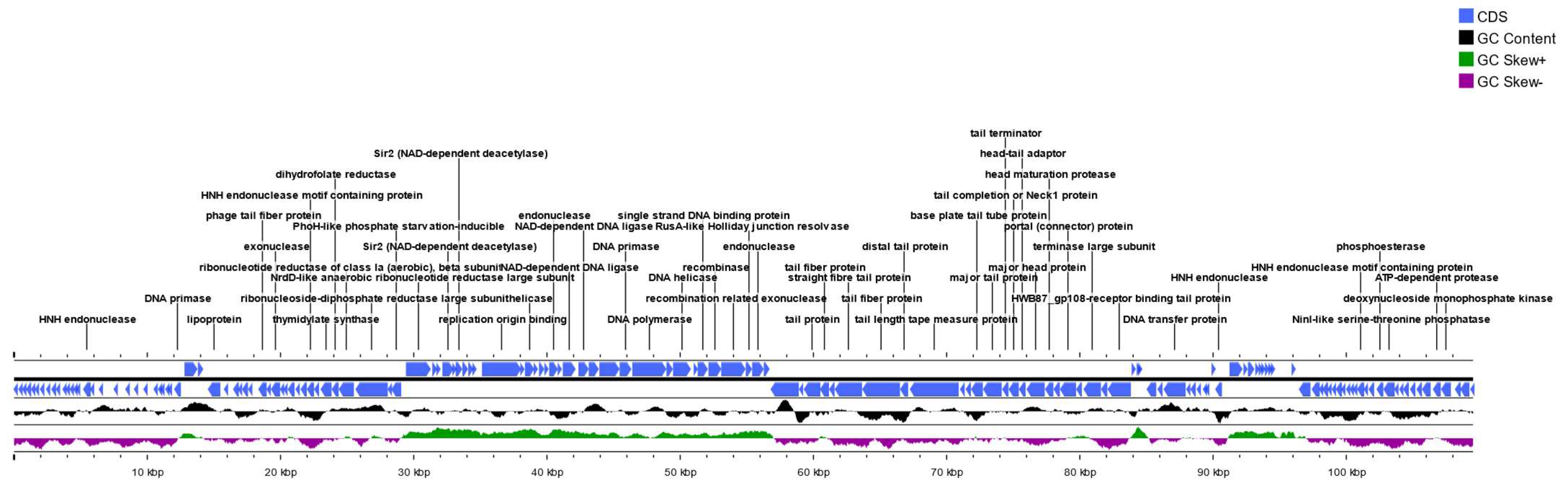
3.2. Annotation, Genome Mapping and Sequence Analysis
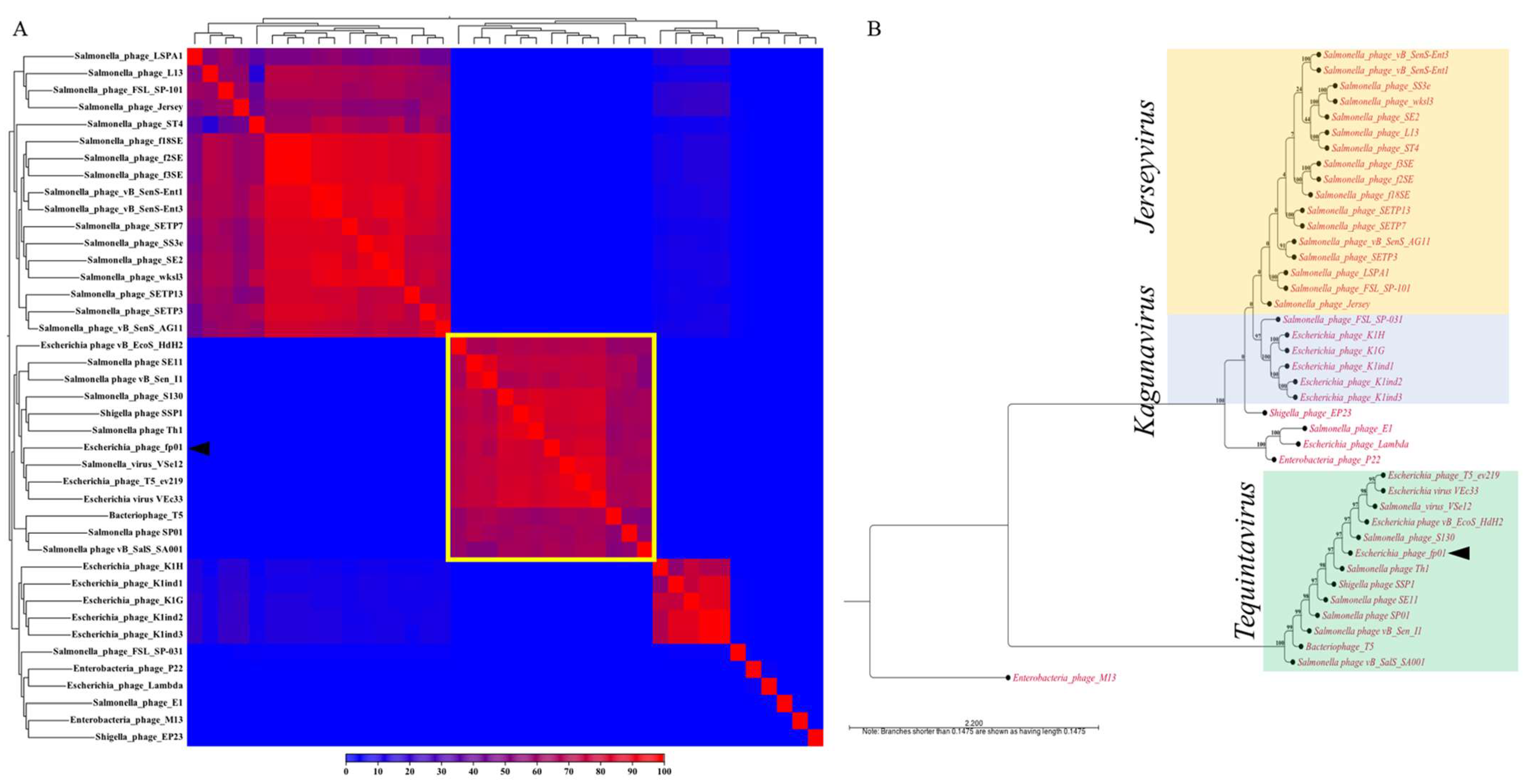
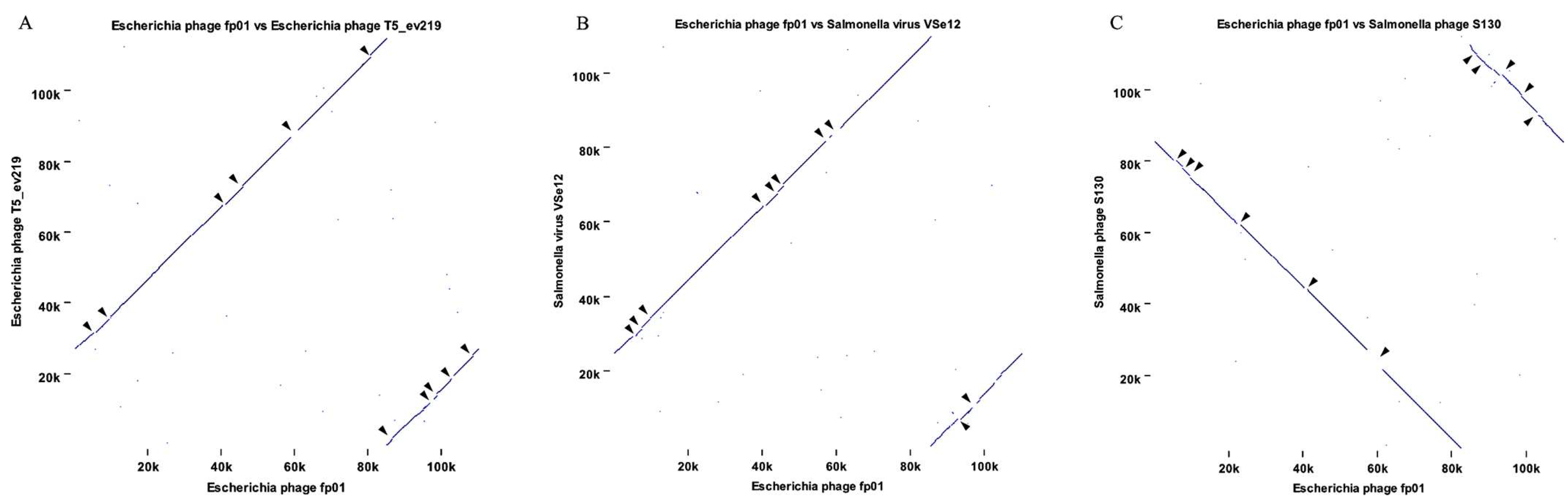
3.3. Comparative Genomics and Phylogenetic Analysis
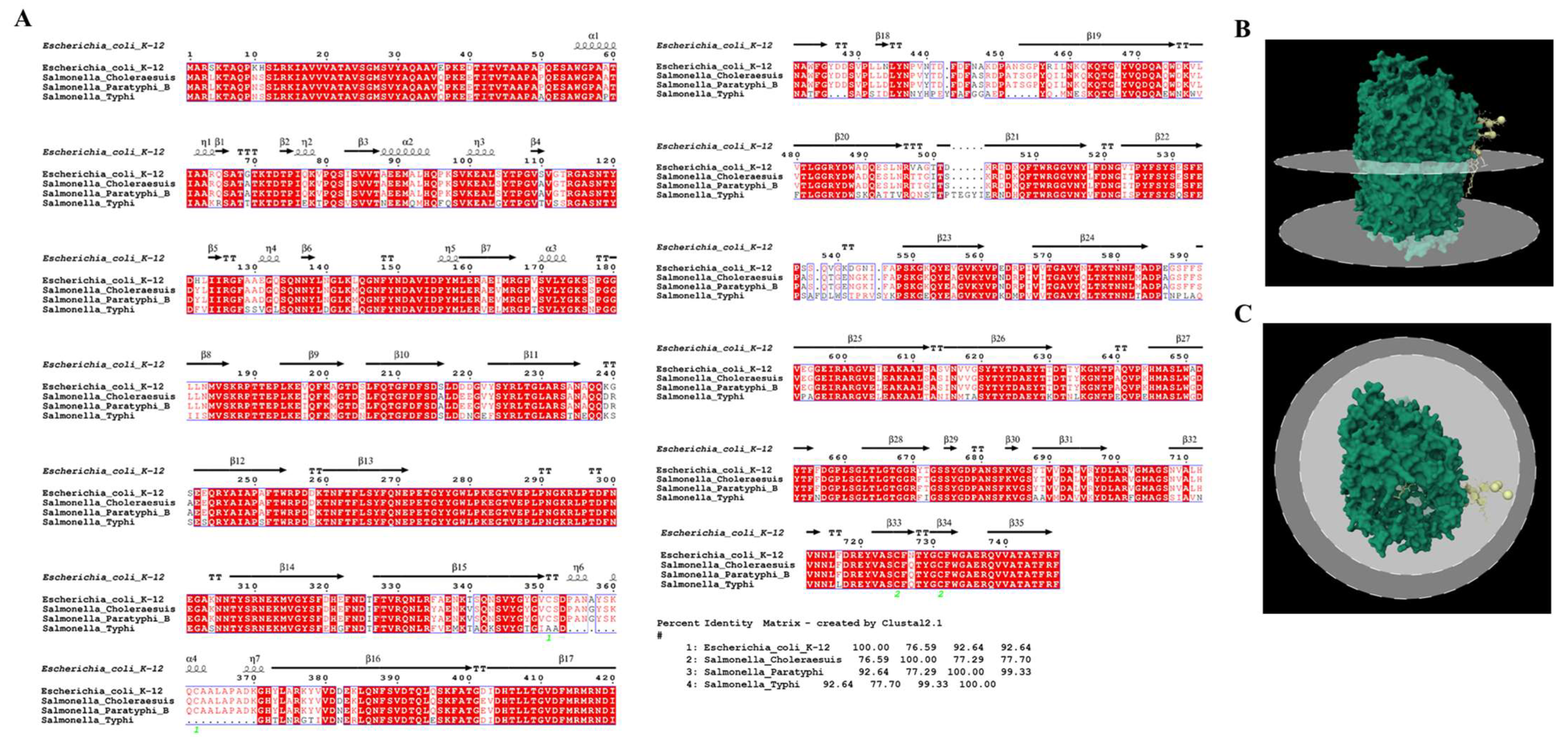
3.4. Receptor Binding Interaction Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Casjens, S.R. Comparative genomics and evolution of the tailed-bacteriophages. Curr. Opin. Microbiol. 2005, 8, 451–458. [Google Scholar] [CrossRef] [PubMed]
- Rohwer, F. Global phage diversity. Cell 2003, 113, 141. [Google Scholar] [CrossRef] [PubMed]
- Sulakvelidze, A.; Alavidze, Z.; Morris, J.G., Jr. Bacteriophage therapy. Antimicrob. Agents Chemother. 2001, 45, 649–659. [Google Scholar] [CrossRef]
- Garcia, P.; Martinez, B.; Obeso, J.M.; Rodriguez, A. Bacteriophages and their application in food safety. Lett. Appl. Microbiol. 2008, 47, 479–485. [Google Scholar] [CrossRef]
- Cooper, C.J.; Khan Mirzaei, M.; Nilsson, A.S. Adapting Drug Approval Pathways for Bacteriophage-Based Therapeutics. Front. Microbiol. 2016, 7, 1209. [Google Scholar] [CrossRef]
- Moye, Z.D.; Woolston, J.; Sulakvelidze, A. Bacteriophage Applications for Food Production and Processing. Viruses 2018, 10, 205. [Google Scholar] [CrossRef]
- Lang, L.H. FDA approves use of bacteriophages to be added to meat and poultry products. Gastroenterology 2006, 131, 1370. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.B.; Vallad, G.E.; Iriarte, F.B.; Obradovic, A.; Wernsing, M.H.; Jackson, L.E.; Balogh, B.; Hong, J.C.; Momol, M.T. Considerations for using bacteriophages for plant disease control. Bacteriophage 2012, 2, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, H.W.; Audurier, A.; Berthiaume, L.; Jones, L.A.; Mayo, J.A.; Vidaver, A.K. Guidelines for bacteriophage characterization. Adv. Virus Res. 1978, 23, 1–24. [Google Scholar] [CrossRef]
- Santander, J.; Robeson, J. Phage-Resistance in Salmonella enterica Serovar Enteritidis and Pathogenicity in Caenorhabditis elegans is Mediate by the Lipopolysaccharide. Electron. J. Biotechnol. 2007, 10, 627–632. [Google Scholar] [CrossRef]
- Hamdi, S.; Rousseau, G.M.; Labrie, S.J.; Tremblay, D.M.; Kourda, R.S.; Ben Slama, K.; Moineau, S. Characterization of two polyvalent phages infecting Enterobacteriaceae. Sci. Rep. 2017, 7, 40349. [Google Scholar] [CrossRef]
- Sarker, S.A.; Sultana, S.; Reuteler, G.; Moine, D.; Descombes, P.; Charton, F.; Bourdin, G.; McCallin, S.; Ngom-Bru, C.; Neville, T.; et al. Oral Phage Therapy of Acute Bacterial Diarrhea With Two Coliphage Preparations: A Randomized Trial in Children From Bangladesh. EBioMedicine 2016, 4, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.K.; Abedon, S.T.; Loc-Carrillo, C. Phage cocktails and the future of phage therapy. Future Microbiol. 2013, 8, 769–783. [Google Scholar] [CrossRef] [PubMed]
- Bronfenbrenner, J. True Polyvalence of Pure Bacteriophages. Proc. Soc. Exp. Biol. Med. 1933, 30, 729–732. [Google Scholar] [CrossRef]
- El Haddad, L.; Ben Abdallah, N.; Plante, P.L.; Dumaresq, J.; Katsarava, R.; Labrie, S.; Corbeil, J.; St-Gelais, D.; Moineau, S. Improving the safety of Staphylococcus aureus polyvalent phages by their production on a Staphylococcus xylosus strain. PLoS ONE 2014, 9, e102600. [Google Scholar] [CrossRef] [PubMed]
- Souza, K.A.; Ginoza, H.S.; Haight, R.D. Isolation of a polyvalent bacteriophage for Escherichia coli, Klebsiella pneumoniae, and Aerobacter aerogenes. J. Virol. 1972, 9, 851–856. [Google Scholar] [CrossRef]
- Yu, P.; Mathieu, J.; Li, M.; Dai, Z.; Alvarez, P.J. Isolation of Polyvalent Bacteriophages by Sequential Multiple-Host Approaches. Appl. Environ. Microbiol. 2016, 82, 808–815. [Google Scholar] [CrossRef]
- Parra, B.; Robeson, J. Selection of polyvalent bacteriophages infecting Salmonella enterica serovar Choleraesuis. Electron. J. Biotechnol. 2016, 21, 72–76. [Google Scholar] [CrossRef]
- Turner, D.; Kropinski, A.M.; Adriaenssens, E.M. A Roadmap for Genome-Based Phage Taxonomy. Viruses 2021, 13, 506. [Google Scholar] [CrossRef]
- Fookes, M.; Schroeder, G.N.; Langridge, G.C.; Blondel, C.J.; Mammina, C.; Connor, T.R.; Seth-Smith, H.; Vernikos, G.S.; Robinson, K.S.; Sanders, M.; et al. Salmonella bongori Provides Insights into the Evolution of the Salmonellae. PLoS Pathog. 2011, 7, e1002191. [Google Scholar] [CrossRef]
- van den Berg, B.; Silale, A.; Baslé, A.; Brandner, A.F.; Mader, S.L.; Khalid, S. Structural basis for host recognition and superinfection exclusion by bacteriophage T5. Proc. Natl. Acad. Sci. USA 2022, 119, e2211672119. [Google Scholar] [CrossRef]
- Ferguson, A.D.; Hofmann, E.; Coulton, J.W.; Diederichs, K.; Welte, W. Siderophore-mediated iron transport: Crystal structure of FhuA with bound lipopolysaccharide. Science 1998, 282, 2215–2220. [Google Scholar] [CrossRef] [PubMed]
- Flayhan, A.; Wien, F.; Paternostre, M.; Boulanger, P.; Breyton, C. New insights into pb5, the receptor binding protein of bacteriophage T5, and its interaction with its Escherichia coli receptor FhuA. Biochimie 2012, 94, 1982–1989. [Google Scholar] [CrossRef]
- Adams, M.H. Bacteriophages; Interscience Publishers: New York, NY, USA, 1959; 592p. [Google Scholar]
- Borie, C.; Albala, I.; Sanchez, P.; Sanchez, M.L.; .Ramirez, S.; .Navarro, C.; .Morales, M.A.; .Retamales, J.; .Robeson, J. Bacteriophage treatment reduces Salmonella colonization of infected chickens. Avian Dis. 2008, 52, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, K.; Murray, N.; Whittaker, P. Construction of representative genomic DNA libraries using phages lambda replacement vectors. In DNA Cloning 1: A Practical Approach; Glover, D., Hames, B., Eds.; Oxford University Press: New York, NY, USA, 1995; pp. 37–83. [Google Scholar]
- Istace, B.; Friedrich, A.; d’Agata, L.; Faye, S.; Payen, E.; Beluche, O.; Caradec, C.; Davidas, S.; Cruaud, C.; Liti, G.; et al. De novo assembly and population genomic survey of natural yeast isolates with the Oxford Nanopore MinION sequencer. Gigascience 2017, 6, giw018. [Google Scholar] [CrossRef] [PubMed]
- Magi, A.; Giusti, B.; Tattini, L. Characterization of MinION nanopore data for resequencing analyses. Brief. Bioinform. 2017, 18, 940–953. [Google Scholar] [CrossRef]
- Magi, A.; Semeraro, R.; Mingrino, A.; Giusti, B.; D’Aurizio, R. Nanopore sequencing data analysis: State of the art, applications and challenges. Brief. Bioinform. 2017, 19, 1256–1272. [Google Scholar] [CrossRef]
- Tyler, A.D.; Mataseje, L.; Urfano, C.J.; Schmidt, L.; Antonation, K.S.; Mulvey, M.R.; Corbett, C.R. Evaluation of Oxford Nanopore’s MinION Sequencing Device for Microbial Whole Genome Sequencing Applications. Sci. Rep. 2018, 8, 10931. [Google Scholar] [CrossRef]
- Lander, E.S.; Waterman, M.S. Genomic mapping by fingerprinting random clones: A mathematical analysis. Genomics 1988, 2, 231–239. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A quality control tool for high throughput sequence data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 December 2022).
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A Fast Phage Search Tool. Nucleic Acids Res. 2011, 39 (Suppl. 2), W347–W352. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar]
- Jukes, T.H.; Cantor, C.R. Evolution of Protein Molecules. In Mammalian Protein Metabolism; Munro, H.N., Ed.; Academic Press: Cambridge, MA, USA, 1969; Chapter 24; pp. 21–132. [Google Scholar]
- Anany, H.; Switt, A.I.; De Lappe, N.; Ackermann, H.W.; Reynolds, D.M.; Kropinski, A.M.; Wiedmann, M.; Griffiths, M.W.; Tremblay, D.; Moineau, S.; et al. A proposed new bacteriophage subfamily: “Jerseyvirinae”. Arch. Virol. 2015, 160, 1021–1033. [Google Scholar] [CrossRef]
- Zeng, W.; Mao, P.; Hong, Y.; Feng, M.; Xu, Z.; Huang, F.; Jing, S. Complete Genome Sequence of the Salmonella enterica Serovar Paratyphi A Bacteriophage LSPA1 Isolated in China. Genome Announc. 2015, 3, e01011–e01014. [Google Scholar] [CrossRef]
- Kang, H.W.; Kim, J.W.; Jung, T.S.; Woo, G.J. wksl3, a New biocontrol agent for Salmonella enterica serovars Enteritidis and Typhimurium in foods: Characterization, application, sequence analysis, and oral acute toxicity study. Appl. Environ. Microbiol. 2013, 79, 1956–1968. [Google Scholar] [CrossRef]
- Santander, J.; Vasquez, J.I.; Segovia, C.; Santos, L.; Turra, G.; Huber, K.; Robeson, J. Complete genome sequence of the Salmonella enterica serovar Enteritidis bacteriophages fSE1C and fSE4C isolated from food matrices. Stand. Genom. Sci. 2017, 12, 1. [Google Scholar] [CrossRef]
- Segovia, C.; Vasquez, I.; Maracaja-Coutinho, V.; Robeson, J.; Santander, J. Complete Genome Sequence of Salmonella enterica Serovar Enteritidis Bacteriophage f18SE, Isolated in Chile. Genome Announc. 2015, 3, e00600-15. [Google Scholar] [CrossRef]
- Bull, J.J.; Vimr, E.; Molineux, I. A tale of tails: Sialidase is key to success in a model of phage therapy against K1-capsulated Escherichia coli. Virology 2010, 398, 79–86. [Google Scholar] [CrossRef]
- Chang, H.W.; Kim, K.H. Comparative genomic analysis of bacteriophage EP23 infecting Shigella sonnei and Escherichia coli. J. Microbiol. 2011, 49, 927–934. [Google Scholar] [CrossRef]
- Pickard, D.; Thomson, N.R.; Baker, S.; Wain, J.; Pardo, M.; Goulding, D.; Hamlin, N.; Choudhary, J.; Threfall, J.; Dougan, G. Molecular characterization of the Salmonella enterica serovar Typhi Vi-typing bacteriophage E1. J. Bacteriol. 2008, 190, 2580–2587. [Google Scholar] [CrossRef]
- Gencay, Y.E.; Gambino, M.; Prüssing, T.F.; Brøndsted, L. The genera of bacteriophages and their receptors are the major determinants of host range. Environ. Microbiol. 2019, 21, 2095–2111. [Google Scholar] [CrossRef]
- Kaliman, A.V.; Kryukov, V.; Bayev, A. The nucleotide sequence of bacteriophage T5 DNA at the region between early and late genes. Nucleic Acids Res. 1988, 16, 6230. [Google Scholar] [CrossRef]
- Pedulla, M.L.; Ford, M.E.; Karthikeyan, T.; Houtz, J.M.; Hendrix, R.W.; Hatfull, G.F.; Poteete, A.R.; Gilcrease, E.B.; Winn-Stapley, D.A.; Casjens, S.R. Corrected sequence of the bacteriophage p22 genome. J. Bacteriol. 2003, 185, 1475–1477. [Google Scholar] [CrossRef]
- Sanger, F.; Coulson, A.R.; Hong, G.F.; Hill, D.F.; Petersen, G.B. Nucleotide sequence of bacteriophage lambda DNA. J. Mol. Biol. 1982, 162, 729–773. [Google Scholar] [CrossRef]
- van Wezenbeek, P.M.; Hulsebos, T.; Schoenmakers, J. Nucleotide sequence of the filamentous bacteriophage M13 DNA genome: Comparison with phage fd. Gene 1980, 11, 129–148. [Google Scholar] [CrossRef]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef]
- Du, Z.; Su, H.; Wang, W.; Ye, L.; Wei, H.; Peng, Z.; Anishchenko, I.; Baker, D.; Yang, J. The trRosetta server for fast and accurate protein structure prediction. Nat. Protoc. 2021, 16, 5634–5651. [Google Scholar] [CrossRef]
- Ko, J.; Park, H.; Heo, L.; Seok, C. GalaxyWEB server for protein structure prediction and refinement. Nucleic Acids Res. 2012, 40, W294–W297. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM database and PPM web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 2012, 40, D370–D376. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Tao, H.; He, J.; Huang, S.Y. The HDOCK server for integrated protein–protein docking. Nat. Protoc. 2020, 15, 1829–1852. [Google Scholar] [CrossRef]
- Xue, L.C.; Rodrigues, J.P.; Kastritis, P.L.; Bonvin, A.M.; Vangone, A. PRODIGY: A web server for predicting the binding affinity of protein–protein complexes. Bioinformatics 2016, 32, 3676–3678. [Google Scholar] [CrossRef] [PubMed]
- Higgins, N.P.; Vologodskii, A.V. Topological Behavior of Plasmid DNA. Microbiol. Spectr. 2015, 3. [Google Scholar] [CrossRef]
- Grenyer, B.; Sperratore, M.; Harrison, M. Bacteriophages and a homology model of a large terminase bacteriophage protein. J. Biotech Res. 2018, 9, 79–89. [Google Scholar]
- Merrill, B.D.; Ward, A.T.; Grose, J.H.; Hope, S. Software-based analysis of bacteriophage genomes, physical ends, and packaging strategies. BMC Genom. 2016, 17, 679. [Google Scholar] [CrossRef]
- Hong, Y.; Pan, Y.; Harman, N.J.; Ebner, P.D. Complete genome sequences of two Escherichia coli O157:H7 phages effective in limiting contamination of food Products. Genome Announc. 2014, 2, e00519-14. [Google Scholar] [CrossRef]
- Feucht, A.; Schmid, A.; Benz, R.; Schwarz, H.; Heller, K.J. Pore formation associated with the tail-tip protein pb2 of bacteriophage T5. J. Biol. Chem. 1990, 265, 18561–18567. [Google Scholar] [CrossRef]
- Hagens, S.; Loessner, M.J. Application of bacteriophages for detection and control of foodborne pathogens. Appl. Microbiol. Biotechnol. 2007, 76, 513–519. [Google Scholar] [CrossRef]
- Kazi, M.; Annapure, U.S. Bacteriophage biocontrol of foodborne pathogens. J. Food Sci. Technol. 2016, 53, 1355–1362. [Google Scholar] [CrossRef]
- de Melo, A.G.; Levesque, S.; Moineau, S. Phages as friends and enemies in food processing. Curr. Opin. Biotechnol. 2018, 49, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Marcó, M.B.; Moineau, S.; Quiberoni, A. Bacteriophages and dairy fermentations. Bacteriophage 2012, 2, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Bueno, E.; García, P.; Martínez, B.; Rodríguez, A. Phage inactivation of Staphylococcus aureus in fresh and hard-type cheeses. Int. J. Food Microbiol. 2012, 158, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Sukumaran, A.T.; Nannapaneni, R.; Kiess, A.; Sharma, C.S. Reduction of Salmonella on chicken breast fillets stored under aerobic or modified atmosphere packaging by the application of lytic bacteriophage preparation SalmoFreshTM. Poult. Sci. 2016, 95, 668–675. [Google Scholar] [CrossRef]
- Sukumaran, A.T.; Nannapaneni, R.; Kiess, A.; Sharma, C.S. Reduction of Salmonella on chicken meat and chicken skin by combined or sequential application of lytic bacteriophage with chemical antimicrobials. Int. J. Food Microbiol. 2015, 207, 8–15. [Google Scholar] [CrossRef]
- Wu, J.-L.; Lin, H.-M.; Jan, L.; Hsu, Y.-L.; Chang, L.-H. Biological control of fish bacterial pathogen, Aeromonas hydrophila, by bacteriophage AH 1. Fish Pathol. 1981, 15, 271–276. [Google Scholar] [CrossRef]
- Richards, G.P. Bacteriophage remediation of bacterial pathogens in aquaculture: A review of the technology. Bacteriophage 2014, 4, e975540. [Google Scholar] [CrossRef]
- Kutter, E.; Sulakvelidze, A. Bacteriophages: Biology and Applications; CRC Press: Boca Raton, FL, USA, 2004. [Google Scholar]
- Ackermann, H.W.; Prangishvili, D. Prokaryote viruses studied by electron microscopy. Arch. Virol. 2012, 157, 1843–1849. [Google Scholar] [CrossRef]
- King, A.M.Q.; Adams, M.J.; Carstens, E.B.; Lefkowitz, E. (Eds.) Part III. The ICTV. In Virus Taxonomy; Elsevier: San Diego, CA, USA, 2012; pp. 1261–1291. Available online: https://www.elsevier.com/books/virus-taxonomy/king/978-0-12-384684-6 (accessed on 1 December 2022).
- Adriaenssens, E.M.; Sullivan, M.B.; Knezevic, P.; van Zyl, L.J.; Sarkar, B.L.; Dutilh, B.E.; Alfenas-Zerbini, P.; Łobocka, M.; Tong, Y.; Brister, J.R.; et al. Taxonomy of prokaryotic viruses: 2018–2019 update from the ICTV Bacterial and Archaeal Viruses Subcommittee. Arch. Virol. 2020, 165, 1253–1260. [Google Scholar] [CrossRef]
- Vander Byl, C.; Kropinski, A.M. Sequence of the genome of Salmonella bacteriophage P22. J. Bacteriol. 2000, 182, 6472–6481. [Google Scholar]
- Casjens, S.R.; Hendrix, R.W. Bacteriophage lambda: Early pioneer and still relevant. Virology 2015, 479–480, 310–330. [Google Scholar] [CrossRef]
- Niu, Y.D.; Liu, H.; Johnson, R.P.; McAllister, T.A.; Stanford, K. Effect of a bacteriophage T5virus on growth of Shiga toxigenic Escherichia coli and Salmonella strains in individual and mixed cultures. Virol. J. 2020, 17, 3. [Google Scholar] [CrossRef]
- Kim, M.; Ryu, S. Characterization of a T5-like coliphage, SPC35, and differential development of resistance to SPC35 in Salmonella enterica serovar Typhimurium and Escherichia coli. Appl. Environ. Microbiol. 2011, 77, 2042–2050. [Google Scholar] [CrossRef]
- Zinke, M.; Schröder, G.; Lange, A. Major tail proteins of bacteriophages of the order Caudovirales. J. Biol. Chem. 2022, 298, 101472. [Google Scholar] [CrossRef]
- Werten, S. Identification of the ssDNA-binding protein of bacteriophage T5: Implications for T5 replication. Bacteriophage 2013, 3, e27304. [Google Scholar] [CrossRef]
- Kaliman, A.V.; Kryukov, V.M.; Bayev, A.A. The nucleotide sequence of the region of bacteriophage T5 early genes D10-D15. Nucleic Acids Res. 1988, 16, 10353–10354. [Google Scholar] [CrossRef]
- Kupczok, A.; Neve, H.; Huang, K.D.; Hoeppner, M.P.; Heller, K.J.; Franz, C.M.A.P.; Dagan, T. Rates of Mutation and Recombination in Siphoviridae Phage Genome Evolution over Three Decades. Mol. Biol. Evol. 2018, 35, 1147–1159. [Google Scholar] [CrossRef]
- Amitai, S.; Yassin, Y.; Engelberg-Kulka, H. MazF-mediated cell death in Escherichia coli: A point of no return. J. Bacteriol. 2004, 186, 8295–8300. [Google Scholar] [CrossRef]
- Berry, J.; Rajaure, M.; Pang, T.; Young, R. The spanin complex is essential for lambda lysis. J. Bacteriol. 2012, 194, 5667–5674. [Google Scholar] [CrossRef]
- Endriß, F.; Braun, V. Loop deletions indicate regions important for FhuA transport and receptor functions in Escherichia coli. J. Bacteriol. 2004, 186, 4818–4823. [Google Scholar] [CrossRef]
- Mondigler, M.; Holz, T.; Heller, K.J. Identification of the receptor-binding regions of pb5 proteins of bacteriophages T5 and BF23. Virology 1996, 219, 19–28. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Family/Subfamily/ Genus | Accession Number | Reference |
|---|---|---|---|
| Salmonella phage SE2 | Jerseyvirus | JQ007353.1 | [37] |
| Salmonella phage ST4 | JX233783.1 | ||
| Salmonella phage vB SenS-Ent2 | HG934469.1 | [37] | |
| Salmonella phage vB SenS-Ent1 | HE775250.1 | [37] | |
| Salmonella phage vB SenS-Ent3 | HG934470.1 | [37] | |
| Salmonella phage SETP3 | EF177456.2 | [37] | |
| Salmonella phage vB SenS AG11 | JX297445.1 | [37] | |
| Salmonella phage SETP13 | KF562864.1 | [37] | |
| Salmonella phage SETP7 | KF562865.1 | [37] | |
| Salmonella phage FSL SP-101 | KC139511.1 | [37] | |
| Salmonella phage LSPA1 | KM272358.1 | [38] | |
| Salmonella phage Jersey | KF148055.1 | ||
| Salmonella phage SS3e | AY730274.2 | [37] | |
| Salmonella phage wksl3 | JX202565.1 | [39] | |
| Salmonella phage fSE1C | KT962832.1 | [40] | |
| Salmonella phage fSE4C | KT881477.1 | [40] | |
| Salmonella phage f18SE | KR270151.1 | [41] | |
| Salmonella phage f2SE | KU951146.1 | Santander Lab | |
| Salmonella phage f3SE | KU951147.1 | Santander Lab | |
| Escherichia phage K1G | Kagunavirus | GU196277.1 | [42] |
| Escherichia phage K1H | GU196278.1 | [42] | |
| Escherichia phage K1ind1 | GU196279.1 | [42] | |
| Escherichia phage K1ind2 | GU196280.1 | [42] | |
| Escherichia phage K1ind3 | GU196281.1 | [42] | |
| Shigella phage EP23 | Dhillonvirus | JN984867.1 | [43] |
| Salmonella phage STsAS | Seoulvirus | MH221128.1 | |
| Salmonella phage FSL SP-031 | Guernseyvirinae | KC139518.1 | [37] |
| Salmonella phage E1 | Macdonaldcampvirus | AM491472.1 | [44] |
| Salmonella phage S130 | Demerecviridae | MH370377.1 | [45] |
| Salmonella phage VSe12 | NC048794.1 | ||
| Bacteriophage T5 | AY543070 | [46] | |
| Escherichia phage T5_ev219 | LR597655.1 | ||
| Escherichia virus VEc33 | NC_048818 | ||
| Escherichia phage vB_EcoS_HdH2 | NC_048748 | ||
| Salmonella phage Th1 | NC_048795 | ||
| Salmonella phage SP01 | NC_047859 | ||
| Salmonella phage SE11 | NC_048786 | ||
| Salmonella phage vB_Sen_l1 | MT233524 | ||
| Salmonella phage vB_SalS_SA001 | MN445182 | ||
| Enterobacteria phage P22 | Lederbergvirus | NC_002371.2 | [47] |
| Enterobacteria phage lambda | Lambdavirus | J02459.1 | [48] |
| Enterobacteria phage M13 | Inoviridae | NC_003287.2 | [49] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasquez, I.; Retamales, J.; Parra, B.; Machimbirike, V.; Robeson, J.; Santander, J. Comparative Genomics of a Polyvalent Escherichia-Salmonella Phage fp01 and In Silico Analysis of Its Receptor Binding Protein and Conserved Enterobacteriaceae Phage Receptor. Viruses 2023, 15, 379. https://doi.org/10.3390/v15020379
Vasquez I, Retamales J, Parra B, Machimbirike V, Robeson J, Santander J. Comparative Genomics of a Polyvalent Escherichia-Salmonella Phage fp01 and In Silico Analysis of Its Receptor Binding Protein and Conserved Enterobacteriaceae Phage Receptor. Viruses. 2023; 15(2):379. https://doi.org/10.3390/v15020379
Chicago/Turabian StyleVasquez, Ignacio, Julio Retamales, Barbara Parra, Vimbai Machimbirike, James Robeson, and Javier Santander. 2023. "Comparative Genomics of a Polyvalent Escherichia-Salmonella Phage fp01 and In Silico Analysis of Its Receptor Binding Protein and Conserved Enterobacteriaceae Phage Receptor" Viruses 15, no. 2: 379. https://doi.org/10.3390/v15020379
APA StyleVasquez, I., Retamales, J., Parra, B., Machimbirike, V., Robeson, J., & Santander, J. (2023). Comparative Genomics of a Polyvalent Escherichia-Salmonella Phage fp01 and In Silico Analysis of Its Receptor Binding Protein and Conserved Enterobacteriaceae Phage Receptor. Viruses, 15(2), 379. https://doi.org/10.3390/v15020379