
Breaking the Silence: Regulation of HIV Transcription and Latency on the Road to a Cure

Abstract
1. Introduction
2. HIV Transcription and Establishment of Latency
3. “Block and Lock” Approach
3.1. RNA-Induced Silencing
3.2. Inhibition of Tat Function
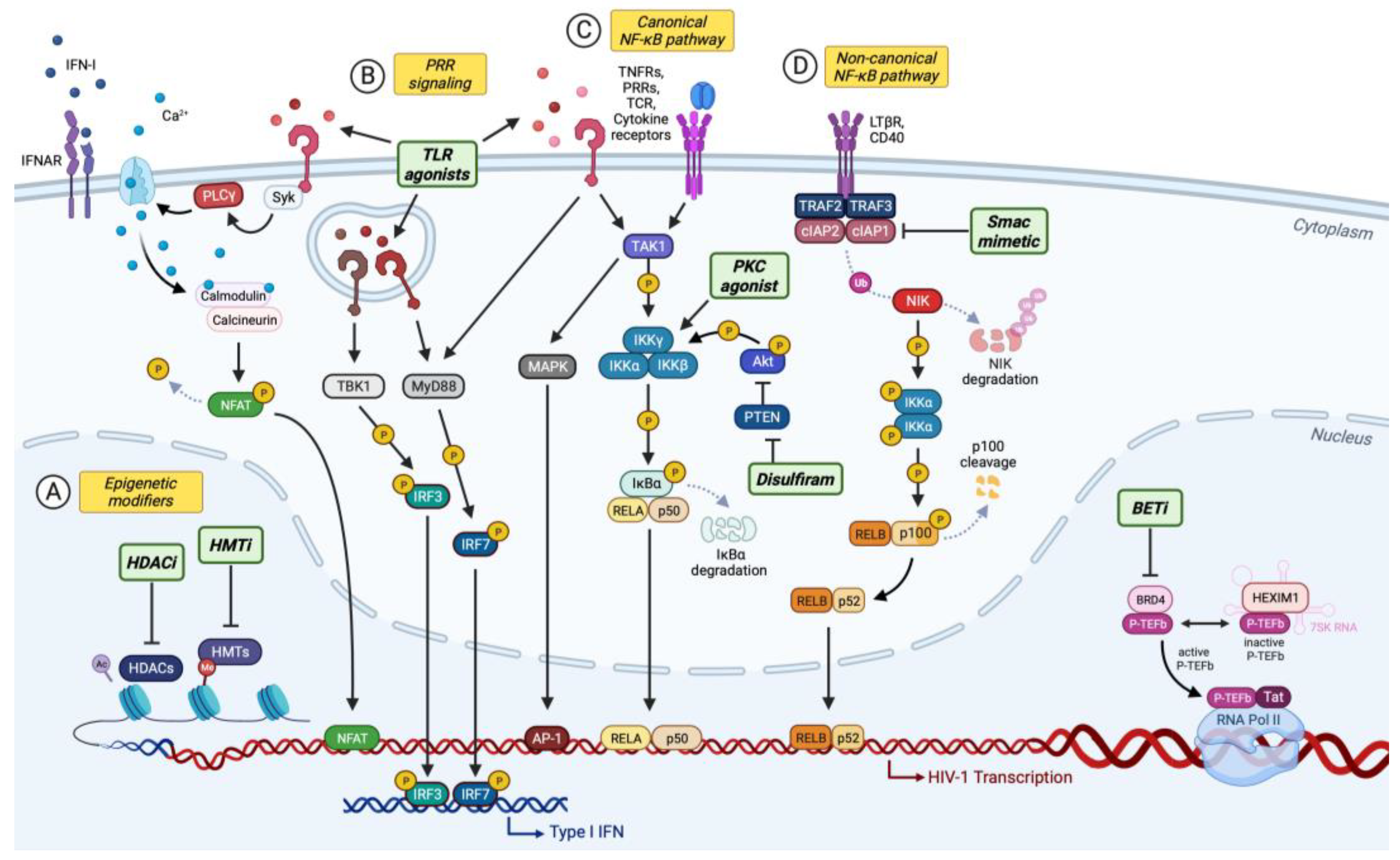
4. “Shock and Kill” Strategy for Curing HIV-1 Infection
4.1. Epigenetic Modifiers
4.1.1. Long Non-Coding RNAs (lncRNAs)
4.1.2. Histone Deacetylase Inhibitors (HDACi)
4.1.3. Histone Methyltransferases (HMT) Inhibitors (HMTi)
4.1.4. Bromodomain and Extra-Terminal Domain Inhibitors (BETi)
4.2. Activators/Inhibitors of Inducible Host Factors
4.2.1. Toll-Like Receptor (TLR) Agonists
4.2.2. Activators of Canonical NF-κB Signaling
4.2.3. Second Mitochondria-Derived Activator of Caspases (Smac) Mimetics
4.3. Latency-Reversing Agents in Combination
4.4. Kill Agents
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Archin, N.M.; Margolis, D.M. Emerging strategies to deplete the HIV reservoir. Curr. Opin. Infect. Dis. 2014, 27, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Zicari, S.; Sessa, L.; Cotugno, N.; Ruggiero, A.; Morrocchi, E.; Concato, C.; Rocca, S.; Zangari, P.; Manno, E.C.; Palma, P. Immune Activation, Inflammation, and Non-AIDS Co-Morbidities in HIV-Infected Patients under Long-Term ART. Viruses 2019, 11, 200. [Google Scholar] [CrossRef]
- Cai, C.W.; Sereti, I. Residual immune dysfunction under antiretroviral therapy. Semin. Immunol. 2021, 51, 101471. [Google Scholar] [CrossRef]
- Alshehri, A.M. Metabolic syndrome and cardiovascular risk. J. Fam. Community Med. 2010, 17, 73–78. [Google Scholar] [CrossRef]
- Bosho, D.D.; Dube, L.; Mega, T.A.; Adare, D.A.; Tesfaye, M.G.; Eshetie, T.C. Prevalence and predictors of metabolic syndrome among people living with human immunodeficiency virus (PLWHIV). Diabetol. Metab. Syndr. 2018, 10, 10. [Google Scholar] [CrossRef]
- Gami, A.S.; Witt, B.J.; Howard, D.E.; Erwin, P.J.; Gami, L.A.; Somers, V.K.; Montori, V.M. Metabolic syndrome and risk of incident cardiovascular events and death: A systematic review and meta-analysis of longitudinal studies. J. Am. Coll. Cardiol. 2007, 49, 403–414. [Google Scholar] [CrossRef]
- Zevin, A.S.; McKinnon, L.; Burgener, A.; Klatt, N.R. Microbial translocation and microbiome dysbiosis in HIV-associated immune activation. Curr. Opin. HIV AIDS 2016, 11, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Raposo, M.A.; Armiliato, G.N.A.; Guimaraes, N.S.; Caram, C.A.; Silveira, R.D.S.; Tupinambas, U. Metabolic disorders and cardiovascular risk in people living with HIV/AIDS without the use of antiretroviral therapy. Rev. Soc. Bras. Med. Trop. 2017, 50, 598–606. [Google Scholar] [CrossRef]
- Deeks, S.G.; Wrin, T.; Liegler, T.; Hoh, R.; Hayden, M.; Barbour, J.D.; Hellmann, N.S.; Petropoulos, C.J.; McCune, J.M.; Hellerstein, M.K.; et al. Virologic and immunologic consequences of discontinuing combination antiretroviral-drug therapy in HIV-infected patients with detectable viremia. N. Engl. J. Med. 2001, 344, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Persaud, D.; Pierson, T.; Ruff, C.; Finzi, D.; Chadwick, K.R.; Margolick, J.B.; Ruff, A.; Hutton, N.; Ray, S.; Siliciano, R.F. A stable latent reservoir for HIV-1 in resting CD4(+) T lymphocytes in infected children. J. Clin. Investig. 2000, 105, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.W.; Stuyver, L.; Mizell, S.B.; Ehler, L.A.; Mican, J.A.; Baseler, M.; Lloyd, A.L.; Nowak, M.A.; Fauci, A.S. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA 1997, 94, 13193–13197. [Google Scholar] [CrossRef] [PubMed]
- Finzi, D.; Blankson, J.; Siliciano, J.D.; Margolick, J.B.; Chadwick, K.; Pierson, T.; Smith, K.; Lisziewicz, J.; Lori, F.; Flexner, C.; et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 1999, 5, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Finzi, D.; Hermankova, M.; Pierson, T.; Carruth, L.M.; Buck, C.; Chaisson, R.E.; Quinn, T.C.; Chadwick, K.; Margolick, J.; Brookmeyer, R.; et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 1997, 278, 1295–1300. [Google Scholar] [CrossRef]
- Barton, K.; Winckelmann, A.; Palmer, S. HIV-1 Reservoirs during Suppressive Therapy. Trends Microbiol. 2016, 24, 345–355. [Google Scholar] [CrossRef]
- Alexaki, A.; Liu, Y.; Wigdahl, B. Cellular reservoirs of HIV-1 and their role in viral persistence. Curr. HIV Res. 2008, 6, 388–400. [Google Scholar] [CrossRef]
- Lutz, C.T.; Karapetyan, A.; Al-Attar, A.; Shelton, B.J.; Holt, K.J.; Tucker, J.H.; Presnell, S.R. Human NK cells proliferate and die in vivo more rapidly than T cells in healthy young and elderly adults. J. Immunol. 2011, 186, 4590–4598. [Google Scholar] [CrossRef]
- Patel, A.A.; Ginhoux, F.; Yona, S. Monocytes, macrophages, dendritic cells and neutrophils: An update on lifespan kinetics in health and disease. Immunology 2021, 163, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Banga, R.; Procopio, F.A.; Lana, E.; Gladkov, G.T.; Roseto, I.; Parsons, E.M.; Lian, X.; Armani-Tourret, M.; Bellefroid, M.; Gao, C.; et al. Lymph node dendritic cells harbor inducible replication-competent HIV despite years of suppressive ART. Cell Host Microbe 2023, 31, 1714–1731. [Google Scholar] [CrossRef]
- Tang, Y.; Chaillon, A.; Gianella, S.; Wong, L.M.; Li, D.; Simermeyer, T.L.; Porrachia, M.; Ignacio, C.; Woodworth, B.; Zhong, D.; et al. Brain microglia serve as a persistent HIV reservoir despite durable antiretroviral therapy. J. Clin. Investig. 2023, 133, e167417. [Google Scholar] [CrossRef]
- Parbie, P.K.; Mizutani, T.; Ishizaka, A.; Kawana-Tachikawa, A.; Runtuwene, L.R.; Seki, S.; Abana, C.Z.; Kushitor, D.; Bonney, E.Y.; Ofori, S.B.; et al. Dysbiotic Fecal Microbiome in HIV-1 Infected Individuals in Ghana. Front. Cell Infect. Microbiol. 2021, 11, 646467. [Google Scholar] [CrossRef]
- Barre-Sinoussi, F.; Ross, A.L.; Delfraissy, J.F. Past, present and future: 30 years of HIV research. Nat. Rev. Microbiol. 2013, 11, 877–883. [Google Scholar] [CrossRef] [PubMed]
- International, A.S.S.W.G.o.H.I.V.C.; Deeks, S.G.; Autran, B.; Berkhout, B.; Benkirane, M.; Cairns, S.; Chomont, N.; Chun, T.W.; Churchill, M.; Di Mascio, M.; et al. Towards an HIV cure: A global scientific strategy. Nat. Rev. Immunol. 2012, 12, 607–614. [Google Scholar] [CrossRef]
- Darcis, G.; Van Driessche, B.; Van Lint, C. HIV Latency: Should We Shock or Lock? Trends Immunol. 2017, 38, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Whitcomb, J.M.; Kumar, R.; Hughes, S.H. Sequence of the circle junction of human immunodeficiency virus type 1: Implications for reverse transcription and integration. J. Virol. 1990, 64, 4903–4906. [Google Scholar] [CrossRef]
- Coffin, J.M.; Hughes, S.H.; Varmus, H.E. (Eds.) Retroviruses; Cold Spring Harbor: New York, NY, USA, 1997. [Google Scholar]
- Schiralli Lester, G.M.; Henderson, A.J. Mechanisms of HIV Transcriptional Regulation and Their Contribution to Latency. Mol. Biol. Int. 2012, 2012, 614120. [Google Scholar] [CrossRef]
- Bannwarth, S.; Gatignol, A. HIV-1 TAR RNA: The target of molecular interactions between the virus and its host. Curr. HIV Res. 2005, 3, 61–71. [Google Scholar] [CrossRef]
- Kilareski, E.M.; Shah, S.; Nonnemacher, M.R.; Wigdahl, B. Regulation of HIV-1 transcription in cells of the monocyte-macrophage lineage. Retrovirology 2009, 6, 118. [Google Scholar] [CrossRef]
- Pereira, L.A.; Bentley, K.; Peeters, A.; Churchill, M.J.; Deacon, N.J. A compilation of cellular transcription factor interactions with the HIV-1 LTR promoter. Nucleic Acids Res. 2000, 28, 663–668. [Google Scholar] [CrossRef]
- Rohr, O.; Marban, C.; Aunis, D.; Schaeffer, E. Regulation of HIV-1 gene transcription: From lymphocytes to microglial cells. J. Leukoc. Biol. 2003, 74, 736–749. [Google Scholar] [CrossRef]
- Li, G.M.Y.; Franza, B.R., Jr. In vitro study of functional involvement of Sp1, NF-kappa B/Rel, and AP1 in phorbol 12-myristate 13-acetate-mediated HIV-1 long terminal repeat activation. J. Biol. Chem. 1994, 269, 30616–30619. [Google Scholar] [CrossRef]
- Majello, B.; De Luca, P.; Hagen, G.; Suske, G.; Lania, L. Different members of the Sp1 multigene family exert opposite transcriptional regulation of the long terminal repeat of HIV-1. Nucleic Acids Res. 1994, 22, 4914–4921. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D.; Edwards, N.L.; Duckett, C.S.; Agranoff, A.B.; Schmid, R.M.; Nabel, G.J. A cooperative interaction between NF-kappa B and Sp1 is required for HIV-1 enhancer activation. EMBO J. 1993, 12, 3551–3558. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Takagi, T.; Wada, T.; Yano, K.; Furuya, A.; Sugimoto, S.; Hasegawa, J.; Handa, H. NELF, a multisubunit complex containing RD, cooperates with DSIF to repress RNA polymerase II elongation. Cell 1999, 97, 41–51. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Margolis, D.M. Counterregulation of chromatin deacetylation and histone deacetylase occupancy at the integrated promoter of human immunodeficiency virus type 1 (HIV-1) by the HIV-1 repressor YY1 and HIV-1 activator Tat. Mol. Cell Biol. 2002, 22, 2965–2973. [Google Scholar] [CrossRef]
- Bernhard, W.; Barreto, K.; Raithatha, S.; Sadowski, I. An upstream YY1 binding site on the HIV-1 LTR contributes to latent infection. PLoS ONE 2013, 8, e77052. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Okamoto, T. Transcriptional repression of human immunodeficiency virus type 1 by AP-4. J. Biol. Chem. 2006, 281, 12495–12505. [Google Scholar] [CrossRef]
- Canonne-Hergaux, F.; Aunis, D.; Schaeffer, E. Interactions of the transcription factor AP-1 with the long terminal repeat of different human immunodeficiency virus type 1 strains in Jurkat, glial, and neuronal cells. J. Virol. 1995, 69, 6634–6642. [Google Scholar] [CrossRef]
- Roebuck, K.A.; Gu, D.S.; Kagnoff, M.F. Activating protein-1 cooperates with phorbol ester activation signals to increase HIV-1 expression. AIDS 1996, 10, 819–826. [Google Scholar] [CrossRef]
- Roebuck, K.A.; Saifuddin, M. Regulation of HIV-1 transcription. Gene Expr. 1999, 8, 67–84. [Google Scholar]
- Schroder, A.R.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef]
- Bushman, F.; Lewinski, M.; Ciuffi, A.; Barr, S.; Leipzig, J.; Hannenhalli, S.; Hoffmann, C. Genome-wide analysis of retroviral DNA integration. Nat. Rev. Microbiol. 2005, 3, 848–858. [Google Scholar] [CrossRef]
- Lewinski, M.K.; Bisgrove, D.; Shinn, P.; Chen, H.; Hoffmann, C.; Hannenhalli, S.; Verdin, E.; Berry, C.C.; Ecker, J.R.; Bushman, F.D. Genome-wide analysis of chromosomal features repressing human immunodeficiency virus transcription. J. Virol. 2005, 79, 6610–6619. [Google Scholar] [CrossRef]
- Lewinski, M.K.; Yamashita, M.; Emerman, M.; Ciuffi, A.; Marshall, H.; Crawford, G.; Collins, F.; Shinn, P.; Leipzig, J.; Hannenhalli, S.; et al. Retroviral DNA integration: Viral and cellular determinants of target-site selection. PLoS Pathog. 2006, 2, e60. [Google Scholar] [CrossRef]
- Duverger, A.; Jones, J.; May, J.; Bibollet-Ruche, F.; Wagner, F.A.; Cron, R.Q.; Kutsch, O. Determinants of the establishment of human immunodeficiency virus type 1 latency. J. Virol. 2009, 83, 3078–3093. [Google Scholar] [CrossRef]
- Jones, K.A.; Kadonaga, J.T.; Luciw, P.A.; Tjian, R. Activation of the AIDS retrovirus promoter by the cellular transcription factor, Sp1. Science 1986, 232, 755–759. [Google Scholar] [CrossRef]
- Shaw, J.P.; Utz, P.J.; Durand, D.B.; Toole, J.J.; Emmel, E.A.; Crabtree, G.R. Identification of a putative regulator of early T cell activation genes. Science 1988, 241, 202–205. [Google Scholar] [CrossRef]
- Osborn, L.; Kunkel, S.; Nabel, G.J. Tumor necrosis factor alpha and interleukin 1 stimulate the human immunodeficiency virus enhancer by activation of the nuclear factor kappa B. Proc. Natl. Acad. Sci. USA 1989, 86, 2336–2340. [Google Scholar] [CrossRef]
- Ni, Z.; Saunders, A.; Fuda, N.J.; Yao, J.; Suarez, J.R.; Webb, W.W.; Lis, J.T. P-TEFb is critical for the maturation of RNA polymerase II into productive elongation in vivo. Mol. Cell Biol. 2008, 28, 1161–1170. [Google Scholar] [CrossRef]
- Ping, Y.H.; Rana, T.M. DSIF and NELF interact with RNA polymerase II elongation complex and HIV-1 Tat stimulates P-TEFb-mediated phosphorylation of RNA polymerase II and DSIF during transcription elongation. J. Biol. Chem. 2001, 276, 12951–12958. [Google Scholar] [CrossRef]
- Peterlin, B.M.; Price, D.H. Controlling the elongation phase of transcription with P-TEFb. Mol. Cell 2006, 23, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Karn, J. The molecular biology of HIV latency: Breaking and restoring the Tat-dependent transcriptional circuit. Curr. Opin. HIV AIDS 2011, 6, 4–11. [Google Scholar] [CrossRef]
- Siliciano, R.F.; Greene, W.C. HIV latency. Cold Spring Harb. Perspect. Med. 2011, 1, a007096. [Google Scholar] [CrossRef] [PubMed]
- Arya, S.K.; Gallo, R.C.; Hahn, B.H.; Shaw, G.M.; Popovic, M.; Salahuddin, S.Z.; Wong-Staal, F. Homology of genome of AIDS-associated virus with genomes of human T-cell leukemia viruses. Science 1984, 225, 927–930. [Google Scholar] [CrossRef] [PubMed]
- Rosen, C.A.; Sodroski, J.G.; Haseltine, W.A. The location of cis-acting regulatory sequences in the human T cell lymphotropic virus type III (HTLV-III/LAV) long terminal repeat. Cell 1985, 41, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Michels, A.A.; Fraldi, A.; Li, Q.; Adamson, T.E.; Bonnet, F.; Nguyen, V.T.; Sedore, S.C.; Price, J.P.; Price, D.H.; Lania, L.; et al. Binding of the 7SK snRNA turns the HEXIM1 protein into a P-TEFb (CDK9/cyclin T) inhibitor. EMBO J. 2004, 23, 2608–2619. [Google Scholar] [CrossRef] [PubMed]
- Dutilleul, A.; Rodari, A.; Van Lint, C. Depicting HIV-1 Transcriptional Mechanisms: A Summary of What We Know. Viruses 2020, 12, 1385. [Google Scholar] [CrossRef] [PubMed]
- Pumfery, A.; Deng, L.; Maddukuri, A.; de la Fuente, C.; Li, H.; Wade, J.D.; Lambert, P.; Kumar, A.; Kashanchi, F. Chromatin remodeling and modification during HIV-1 Tat-activated transcription. Curr. HIV Res. 2003, 1, 343–362. [Google Scholar] [CrossRef]
- Machida, S.; Depierre, D.; Chen, H.C.; Thenin-Houssier, S.; Petitjean, G.; Doyen, C.M.; Takaku, M.; Cuvier, O.; Benkirane, M. Exploring histone loading on HIV DNA reveals a dynamic nucleosome positioning between unintegrated and integrated viral genome. Proc. Natl. Acad. Sci. USA 2020, 117, 6822–6830. [Google Scholar] [CrossRef]
- Rafati, H.; Parra, M.; Hakre, S.; Moshkin, Y.; Verdin, E.; Mahmoudi, T. Repressive LTR nucleosome positioning by the BAF complex is required for HIV latency. PLoS Biol. 2011, 9, e1001206. [Google Scholar] [CrossRef]
- Boekhoudt, G.H.; Guo, Z.; Beresford, G.W.; Boss, J.M. Communication between NF-kappa B and Sp1 controls histone acetylation within the proximal promoter of the monocyte chemoattractant protein 1 gene. J. Immunol. 2003, 170, 4139–4147. [Google Scholar] [CrossRef]
- Bhatt, D.; Ghosh, S. Regulation of the NF-kappaB-Mediated Transcription of Inflammatory Genes. Front. Immunol. 2014, 5, 71. [Google Scholar] [CrossRef] [PubMed]
- Petty, E.; Pillus, L. Balancing chromatin remodeling and histone modifications in transcription. Trends Genet. 2013, 29, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Ajasin, D.; Eugenin, E.A. HIV-1 Tat: Role in Bystander Toxicity. Front. Cell Infect. Microbiol. 2020, 10, 61. [Google Scholar] [CrossRef]
- Besnard, E.; Hakre, S.; Kampmann, M.; Lim, H.W.; Hosmane, N.N.; Martin, A.; Bassik, M.C.; Verschueren, E.; Battivelli, E.; Chan, J.; et al. The mTOR Complex Controls HIV Latency. Cell Host Microbe 2016, 20, 785–797. [Google Scholar] [CrossRef]
- Pasquereau, S.; Herbein, G. CounterAKTing HIV: Toward a “Block and Clear” Strategy? Front. Cell Infect. Microbiol. 2022, 12, 827717. [Google Scholar] [CrossRef]
- Cary, D.C.; Fujinaga, K.; Peterlin, B.M. Molecular mechanisms of HIV latency. J. Clin. Investig. 2016, 126, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Ahlenstiel, C.L.; Symonds, G.; Kent, S.J.; Kelleher, A.D. Block and Lock HIV Cure Strategies to Control the Latent Reservoir. Front. Cell Infect. Microbiol. 2020, 10, 424. [Google Scholar] [CrossRef]
- Li, C.; Mori, L.P.; Lyu, S.; Bronson, R.; Getzler, A.J.; Pipkin, M.E.; Valente, S.T. The chaperone protein p32 stabilizes HIV-1 Tat and strengthens the p-TEFb/RNAPII/TAR complex promoting HIV transcription elongation. Proc. Natl. Acad. Sci. USA 2023, 120, e2217476120. [Google Scholar] [CrossRef]
- Mendez, C.; Ledger, S.; Petoumenos, K.; Ahlenstiel, C.; Kelleher, A.D. RNA-induced epigenetic silencing inhibits HIV-1 reactivation from latency. Retrovirology 2018, 15, 67. [Google Scholar] [CrossRef]
- Vansant, G.; Bruggemans, A.; Janssens, J.; Debyser, Z. Block-And-Lock Strategies to Cure HIV Infection. Viruses 2020, 12, 84. [Google Scholar] [CrossRef]
- Surabhi, R.M.; Gaynor, R.B. RNA interference directed against viral and cellular targets inhibits human immunodeficiency Virus Type 1 replication. J. Virol. 2002, 76, 12963–12973. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Shijuuku, T.; Fukamachi, T.; Zaunders, J.; Guillemin, G.; Cooper, D.; Kelleher, A. Prolonged transcriptional silencing and CpG methylation induced by siRNAs targeted to the HIV-1 promoter region. J. RNAi Gene Silencing 2005, 1, 66–78. [Google Scholar] [PubMed]
- Suzuki, K.; Ishida, T.; Yamagishi, M.; Ahlenstiel, C.; Swaminathan, S.; Marks, K.; Murray, D.; McCartney, E.M.; Beard, M.R.; Alexander, M.; et al. Transcriptional gene silencing of HIV-1 through promoter targeted RNA is highly specific. RNA Biol. 2011, 8, 1035–1046. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Ahlenstiel, C.; Marks, K.; Kelleher, A.D. Promoter Targeting RNAs: Unexpected Contributors to the Control of HIV-1 Transcription. Mol. Ther. Nucleic Acids 2015, 4, e222. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, T. Transcriptional gene silencing limits CXCR4-associated depletion of bone marrow CD34+ cells in HIV-1 infection. AIDS 2018, 32, 1737–1747. [Google Scholar] [CrossRef]
- Yamagishi, M.; Ishida, T.; Miyake, A.; Cooper, D.A.; Kelleher, A.D.; Suzuki, K.; Watanabe, T. Retroviral delivery of promoter-targeted shRNA induces long-term silencing of HIV-1 transcription. Microbes Infect. 2009, 11, 500–508. [Google Scholar] [CrossRef]
- Weinberg, M.S.; Villeneuve, L.M.; Ehsani, A.; Amarzguioui, M.; Aagaard, L.; Chen, Z.X.; Riggs, A.D.; Rossi, J.J.; Morris, K.V. The antisense strand of small interfering RNAs directs histone methylation and transcriptional gene silencing in human cells. RNA 2006, 12, 256–262. [Google Scholar] [CrossRef]
- Singh, A.; Palanichamy, J.K.; Ramalingam, P.; Kassab, M.A.; Bhagat, M.; Andrabi, R.; Luthra, K.; Sinha, S.; Chattopadhyay, P. Long-term suppression of HIV-1C virus production in human peripheral blood mononuclear cells by LTR heterochromatization with a short double-stranded RNA. J. Antimicrob. Chemother. 2014, 69, 404–415. [Google Scholar] [CrossRef]
- Hu, B.; Zhong, L.; Weng, Y.; Peng, L.; Huang, Y.; Zhao, Y.; Liang, X.J. Therapeutic siRNA: State of the art. Signal Transduct. Target. Ther. 2020, 5, 101. [Google Scholar] [CrossRef]
- Martinez, M.A. Progress in the therapeutic applications of siRNAs against HIV-1. Methods Mol. Biol. 2009, 487, 343–368. [Google Scholar] [CrossRef]
- Ray, R.M.; Morris, K.V. Long Non-coding RNAs Mechanisms of Action in HIV-1 Modulation and the Identification of Novel Therapeutic Targets. Noncoding RNA 2020, 6, 12. [Google Scholar] [CrossRef]
- Kopp, F.; Mendell, J.T. Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell 2018, 172, 393–407. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, P.W.; Pantoja, C.; Beliakova-Bethell, N. An Evaluation on the Role of Non-Coding RNA in HIV Transcription and Latency: A Review. HIV AIDS 2023, 15, 115–134. [Google Scholar] [CrossRef] [PubMed]
- Saayman, S.; Ackley, A.; Turner, A.W.; Famiglietti, M.; Bosque, A.; Clemson, M.; Planelles, V.; Morris, K.V. An HIV-encoded antisense long noncoding RNA epigenetically regulates viral transcription. Mol. Ther. 2014, 22, 1164–1175. [Google Scholar] [CrossRef] [PubMed]
- Imam, H.; Bano, A.S.; Patel, P.; Holla, P.; Jameel, S. The lncRNA NRON modulates HIV-1 replication in a NFAT-dependent manner and is differentially regulated by early and late viral proteins. Sci. Rep. 2015, 5, 8639. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi-Ishihara, M.; Yamagishi, M.; Hara, T.; Matsuda, Y.; Takahashi, R.; Miyake, A.; Nakano, K.; Yamochi, T.; Ishida, T.; Watanabe, T. HIV-1-encoded antisense RNA suppresses viral replication for a prolonged period. Retrovirology 2012, 9, 38. [Google Scholar] [CrossRef]
- Li, H.; Chi, X.; Li, R.; Ouyang, J.; Chen, Y. A Novel lncRNA, AK130181, Contributes to HIV-1 Latency by Regulating Viral Promoter-Driven Gene Expression in Primary CD4(+) T Cells. Mol. Ther. Nucleic Acids 2020, 20, 754–763. [Google Scholar] [CrossRef]
- Li, J.; Chen, C.; Ma, X.; Geng, G.; Liu, B.; Zhang, Y.; Zhang, S.; Zhong, F.; Liu, C.; Yin, Y.; et al. Long noncoding RNA NRON contributes to HIV-1 latency by specifically inducing tat protein degradation. Nat. Commun. 2016, 7, 11730. [Google Scholar] [CrossRef] [PubMed]
- Trypsteen, W.; White, C.H.; Mukim, A.; Spina, C.A.; De Spiegelaere, W.; Lefever, S.; Planelles, V.; Bosque, A.; Woelk, C.H.; Vandekerckhove, L.; et al. Long non-coding RNAs and latent HIV—A search for novel targets for latency reversal. PLoS ONE 2019, 14, e0224879. [Google Scholar] [CrossRef]
- Wang, H.; Liu, Y.; Huan, C.; Yang, J.; Li, Z.; Zheng, B.; Wang, Y.; Zhang, W. NF-kappaB-Interacting Long Noncoding RNA Regulates HIV-1 Replication and Latency by Repressing NF-kappaB Signaling. J. Virol. 2020, 94, e01057-20. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, C.Y.; Yedavalli, V.S.; Jeang, K.T. NEAT1 long noncoding RNA and paraspeckle bodies modulate HIV-1 posttranscriptional expression. mBio 2013, 4, e00596-12. [Google Scholar] [CrossRef]
- Rice, A.P. The HIV-1 Tat Protein: Mechanism of Action and Target for HIV-1 Cure Strategies. Curr. Pharm. Des. 2017, 23, 4098–4102. [Google Scholar] [CrossRef]
- Nekhai, S.; Jeang, K.T. Transcriptional and post-transcriptional regulation of HIV-1 gene expression: Role of cellular factors for Tat and Rev. Future Microbiol. 2006, 1, 417–426. [Google Scholar] [CrossRef]
- Vardabasso, C.; Manganaro, L.; Lusic, M.; Marcello, A.; Giacca, M. The histone chaperone protein Nucleosome Assembly Protein-1 (hNAP-1) binds HIV-1 Tat and promotes viral transcription. Retrovirology 2008, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Meredith, L.W.; Sivakumaran, H.; Major, L.; Suhrbier, A.; Harrich, D. Potent inhibition of HIV-1 replication by a Tat mutant. PLoS ONE 2009, 4, e7769. [Google Scholar] [CrossRef]
- Lin, M.H.; Sivakumaran, H.; Apolloni, A.; Wei, T.; Jans, D.A.; Harrich, D. Nullbasic, a potent anti-HIV tat mutant, induces CRM1-dependent disruption of HIV rev trafficking. PLoS ONE 2012, 7, e51466. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Li, D.; Sivakumaran, H.; Lor, M.; Rustanti, L.; Cloonan, N.; Wani, S.; Harrich, D. Shutdown of HIV-1 Transcription in T Cells by Nullbasic, a Mutant Tat Protein. mBio 2016, 7, e00518-16. [Google Scholar] [CrossRef]
- Jin, H.; Sun, Y.; Li, D.; Lin, M.H.; Lor, M.; Rustanti, L.; Harrich, D. Strong In Vivo Inhibition of HIV-1 Replication by Nullbasic, a Tat Mutant. mBio 2019, 10, e01769-19. [Google Scholar] [CrossRef] [PubMed]
- Mousseau, G.; Valente, S.T. Didehydro-Cortistatin A: A new player in HIV-therapy? Expert. Rev. Anti Infect. Ther. 2016, 14, 145–148. [Google Scholar] [CrossRef]
- Mousseau, G.; Aneja, R.; Clementz, M.A.; Mediouni, S.; Lima, N.S.; Haregot, A.; Kessing, C.F.; Jablonski, J.A.; Thenin-Houssier, S.; Nagarsheth, N.; et al. Resistance to the Tat Inhibitor Didehydro-Cortistatin A Is Mediated by Heightened Basal HIV-1 Transcription. mBio 2019, 10, e01750-18. [Google Scholar] [CrossRef]
- Mousseau, G.; Kessing, C.F.; Fromentin, R.; Trautmann, L.; Chomont, N.; Valente, S.T. The Tat Inhibitor Didehydro-Cortistatin A Prevents HIV-1 Reactivation from Latency. mBio 2015, 6, e00465. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Mousseau, G.; Valente, S.T. Tat inhibition by didehydro-Cortistatin A promotes heterochromatin formation at the HIV-1 long terminal repeat. Epigenetics Chromatin 2019, 12, 23. [Google Scholar] [CrossRef]
- Kessing, C.F.; Nixon, C.C.; Li, C.; Tsai, P.; Takata, H.; Mousseau, G.; Ho, P.T.; Honeycutt, J.B.; Fallahi, M.; Trautmann, L.; et al. In Vivo Suppression of HIV Rebound by Didehydro-Cortistatin A, a “Block-and-Lock” Strategy for HIV-1 Treatment. Cell Rep. 2017, 21, 600–611. [Google Scholar] [CrossRef] [PubMed]
- Mediouni, S.; Chinthalapudi, K.; Ekka, M.K.; Usui, I.; Jablonski, J.A.; Clementz, M.A.; Mousseau, G.; Nowak, J.; Macherla, V.R.; Beverage, J.N.; et al. Didehydro-Cortistatin A Inhibits HIV-1 by Specifically Binding to the Unstructured Basic Region of Tat. mBio 2019, 10, e02662-18. [Google Scholar] [CrossRef] [PubMed]
- Siliciano, J.D.; Siliciano, R.F. Recent developments in the search for a cure for HIV-1 infection: Targeting the latent reservoir for HIV-1. J. Allergy Clin. Immunol. 2014, 134, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Archin, N.M.; Liberty, A.L.; Kashuba, A.D.; Choudhary, S.K.; Kuruc, J.D.; Crooks, A.M.; Parker, D.C.; Anderson, E.M.; Kearney, M.F.; Strain, M.C.; et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 2012, 487, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Gay, C.L.; James, K.S.; Tuyishime, M.; Falcinelli, S.D.; Joseph, S.B.; Moeser, M.J.; Allard, B.; Kirchherr, J.L.; Clohosey, M.; Raines, S.L.M.; et al. Stable Latent HIV Infection and Low-level Viremia Despite Treatment with the Broadly Neutralizing Antibody VRC07-523LS and the Latency Reversal Agent Vorinostat. J. Infect. Dis. 2022, 225, 856–861. [Google Scholar] [CrossRef] [PubMed]
- Gruell, H.; Gunst, J.D.; Cohen, Y.Z.; Pahus, M.H.; Malin, J.J.; Platten, M.; Millard, K.G.; Tolstrup, M.; Jones, R.B.; Conce Alberto, W.D.; et al. Effect of 3BNC117 and romidepsin on the HIV-1 reservoir in people taking suppressive antiretroviral therapy (ROADMAP): A randomised, open-label, phase 2A trial. Lancet Microbe 2022, 3, e203–e214. [Google Scholar] [CrossRef]
- Gutierrez, C.; Serrano-Villar, S.; Madrid-Elena, N.; Perez-Elias, M.J.; Martin, M.E.; Barbas, C.; Ruiperez, J.; Munoz, E.; Munoz-Fernandez, M.A.; Castor, T.; et al. Bryostatin-1 for latent virus reactivation in HIV-infected patients on antiretroviral therapy. AIDS 2016, 30, 1385–1392. [Google Scholar] [CrossRef]
- Kroon, E.; Ananworanich, J.; Pagliuzza, A.; Rhodes, A.; Phanuphak, N.; Trautmann, L.; Mitchell, J.L.; Chintanaphol, M.; Intasan, J.; Pinyakorn, S.; et al. A randomized trial of vorinostat with treatment interruption after initiating antiretroviral therapy during acute HIV-1 infection. J. Virus Erad. 2020, 6, 100004. [Google Scholar] [CrossRef]
- McMahon, D.K.; Zheng, L.; Cyktor, J.C.; Aga, E.; Macatangay, B.J.; Godfrey, C.; Para, M.; Mitsuyasu, R.T.; Hesselgesser, J.; Dragavon, J.; et al. A Phase 1/2 Randomized, Placebo-Controlled Trial of Romidespin in Persons with HIV-1 on Suppressive Antiretroviral Therapy. J. Infect. Dis. 2021, 224, 648–656. [Google Scholar] [CrossRef]
- Rasmussen, T.A.; Tolstrup, M.; Brinkmann, C.R.; Olesen, R.; Erikstrup, C.; Solomon, A.; Winckelmann, A.; Palmer, S.; Dinarello, C.; Buzon, M.; et al. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: A phase 1/2, single group, clinical trial. Lancet HIV 2014, 1, e13–e21. [Google Scholar] [CrossRef] [PubMed]
- Riddler, S. Vesatolimod (GS-9620) is Safe and Pharmacodynamically Active in HIV-Infected Individuals. In Proceedings of the International AIDS Society Conference on HIV Science, Mexico City, Mexico, 21–24 July 2019. [Google Scholar]
- Sogaard, O.S.; Graversen, M.E.; Leth, S.; Olesen, R.; Brinkmann, C.R.; Nissen, S.K.; Kjaer, A.S.; Schleimann, M.H.; Denton, P.W.; Hey-Cunningham, W.J.; et al. The Depsipeptide Romidepsin Reverses HIV-1 Latency In Vivo. PLoS Pathog. 2015, 11, e1005142. [Google Scholar] [CrossRef]
- Vibholm, L.; Schleimann, M.H.; Hojen, J.F.; Benfield, T.; Offersen, R.; Rasmussen, K.; Olesen, R.; Dige, A.; Agnholt, J.; Grau, J.; et al. Short-Course Toll-Like Receptor 9 Agonist Treatment Impacts Innate Immunity and Plasma Viremia in Individuals With Human Immunodeficiency Virus Infection. Clin. Infect. Dis. 2017, 64, 1686–1695. [Google Scholar] [CrossRef] [PubMed]
- Vibholm, L.K.; Konrad, C.V.; Schleimann, M.H.; Frattari, G.; Winckelmann, A.; Klastrup, V.; Jensen, N.M.; Jensen, S.S.; Schmidt, M.; Wittig, B.; et al. Effects of 24-week Toll-like receptor 9 agonist treatment in HIV type 1+ individuals. AIDS 2019, 33, 1315–1325. [Google Scholar] [CrossRef] [PubMed]
- Dahabieh, M.S.; Battivelli, E.; Verdin, E. Understanding HIV latency: The road to an HIV cure. Annu. Rev. Med. 2015, 66, 407–421. [Google Scholar] [CrossRef]
- Marsden, M.D.; Loy, B.A.; Wu, X.; Ramirez, C.M.; Schrier, A.J.; Murray, D.; Shimizu, A.; Ryckbosch, S.M.; Near, K.E.; Chun, T.W.; et al. In vivo activation of latent HIV with a synthetic bryostatin analog effects both latent cell “kick” and “kill” in strategy for virus eradication. PLoS Pathog. 2017, 13, e1006575. [Google Scholar] [CrossRef]
- Chao, T.C.; Zhang, Q.; Li, Z.; Tiwari, S.K.; Qin, Y.; Yau, E.; Sanchez, A.; Singh, G.; Chang, K.; Kaul, M.; et al. The Long Noncoding RNA HEAL Regulates HIV-1 Replication through Epigenetic Regulation of the HIV-1 Promoter. mBio 2019, 10, e02016-19. [Google Scholar] [CrossRef]
- Qu, D.; Sun, W.W.; Li, L.; Ma, L.; Sun, L.; Jin, X.; Li, T.; Hou, W.; Wang, J.H. Long noncoding RNA MALAT1 releases epigenetic silencing of HIV-1 replication by displacing the polycomb repressive complex 2 from binding to the LTR promoter. Nucleic Acids Res. 2019, 47, 3013–3027. [Google Scholar] [CrossRef]
- Taunton, J.; Hassig, C.A.; Schreiber, S.L. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science 1996, 272, 408–411. [Google Scholar] [CrossRef]
- Yang, W.M.; Inouye, C.; Zeng, Y.; Bearss, D.; Seto, E. Transcriptional repression by YY1 is mediated by interaction with a mammalian homolog of the yeast global regulator RPD3. Proc. Natl. Acad. Sci. USA 1996, 93, 12845–12850. [Google Scholar] [CrossRef]
- Hu, E.; Chen, Z.; Fredrickson, T.; Zhu, Y.; Kirkpatrick, R.; Zhang, G.F.; Johanson, K.; Sung, C.M.; Liu, R.; Winkler, J. Cloning and characterization of a novel human class I histone deacetylase that functions as a transcription repressor. J. Biol. Chem. 2000, 275, 15254–15264. [Google Scholar] [CrossRef] [PubMed]
- Grozinger, C.M.; Hassig, C.A.; Schreiber, S.L. Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc. Natl. Acad. Sci. USA 1999, 96, 4868–4873. [Google Scholar] [CrossRef] [PubMed]
- Kao, H.Y.; Downes, M.; Ordentlich, P.; Evans, R.M. Isolation of a novel histone deacetylase reveals that class I and class II deacetylases promote SMRT-mediated repression. Genes. Dev. 2000, 14, 55–66. [Google Scholar] [CrossRef]
- Zhou, X.; Richon, V.M.; Rifkind, R.A.; Marks, P.A. Identification of a transcriptional repressor related to the noncatalytic domain of histone deacetylases 4 and 5. Proc. Natl. Acad. Sci. USA 2000, 97, 1056–1061. [Google Scholar] [CrossRef]
- Kao, H.Y.; Lee, C.H.; Komarov, A.; Han, C.C.; Evans, R.M. Isolation and characterization of mammalian HDAC10, a novel histone deacetylase. J. Biol. Chem. 2002, 277, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Cueto, M.A.; Asselbergs, F.; Atadja, P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J. Biol. Chem. 2002, 277, 25748–25755. [Google Scholar] [CrossRef]
- Glozak, M.A.; Seto, E. Acetylation/deacetylation modulates the stability of DNA replication licensing factor Cdt1. J. Biol. Chem. 2009, 284, 11446–11453. [Google Scholar] [CrossRef]
- Brachmann, C.B.; Sherman, J.M.; Devine, S.E.; Cameron, E.E.; Pillus, L.; Boeke, J.D. The SIR2 gene family, conserved from bacteria to humans, functions in silencing, cell cycle progression, and chromosome stability. Genes. Dev. 1995, 9, 2888–2902. [Google Scholar] [CrossRef]
- Frye, R.A. Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem. Biophys. Res. Commun. 1999, 260, 273–279. [Google Scholar] [CrossRef]
- Du, J.; Zhou, Y.; Su, X.; Yu, J.J.; Khan, S.; Jiang, H.; Kim, J.; Woo, J.; Kim, J.H.; Choi, B.H.; et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 2011, 334, 806–809. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [PubMed]
- Zaikos, T.D.; Painter, M.M.; Sebastian Kettinger, N.T.; Terry, V.H.; Collins, K.L. Class 1-Selective Histone Deacetylase (HDAC) Inhibitors Enhance HIV Latency Reversal while Preserving the Activity of HDAC Isoforms Necessary for Maximal HIV Gene Expression. J. Virol. 2018, 92, e02110-17. [Google Scholar] [CrossRef] [PubMed]
- Matalon, S.; Rasmussen, T.A.; Dinarello, C.A. Histone deacetylase inhibitors for purging HIV-1 from the latent reservoir. Mol. Med. 2011, 17, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Zhang, Y.; Lin, S.; Han, Y.; Zhu, H.Z. Histone deacetylase inhibitor Scriptaid reactivates latent HIV-1 promoter by inducing histone modification in in vitro latency cell lines. Int. J. Mol. Med. 2010, 26, 265–272. [Google Scholar] [CrossRef]
- Yin, H.; Zhang, Y.; Zhou, X.; Zhu, H. Histonedeacetylase inhibitor Oxamflatin increase HIV-1 transcription by inducing histone modification in latently infected cells. Mol. Biol. Rep. 2011, 38, 5071–5078. [Google Scholar] [CrossRef]
- Mates, J.M.; de Silva, S.; Lustberg, M.; Van Deusen, K.; Baiocchi, R.A.; Wu, L.; Kwiek, J.J. A Novel Histone Deacetylase Inhibitor, AR-42, Reactivates HIV-1 from Chronically and Latently Infected CD4(+) T-cells. Retrovirology 2015, 7, 1–5. [Google Scholar] [CrossRef][Green Version]
- Kiernan, R.E.; Vanhulle, C.; Schiltz, L.; Adam, E.; Xiao, H.; Maudoux, F.; Calomme, C.; Burny, A.; Nakatani, Y.; Jeang, K.T.; et al. HIV-1 tat transcriptional activity is regulated by acetylation. EMBO J. 1999, 18, 6106–6118. [Google Scholar] [CrossRef]
- Divsalar, D.N.; Simoben, C.V.; Schonhofer, C.; Richard, K.; Sippl, W.; Ntie-Kang, F.; Tietjen, I. Novel Histone Deacetylase Inhibitors and HIV-1 Latency-Reversing Agents Identified by Large-Scale Virtual Screening. Front. Pharmacol. 2020, 11, 905. [Google Scholar] [CrossRef]
- Spina, C.A.; Anderson, J.; Archin, N.M.; Bosque, A.; Chan, J.; Famiglietti, M.; Greene, W.C.; Kashuba, A.; Lewin, S.R.; Margolis, D.M.; et al. An in-depth comparison of latent HIV-1 reactivation in multiple cell model systems and resting CD4+ T cells from aviremic patients. PLoS Pathog. 2013, 9, e1003834. [Google Scholar] [CrossRef]
- Boateng, A.T.; Abaidoo-Myles, A.; Bonney, E.Y.; Kyei, G.B. Isoform-Selective Versus Nonselective Histone Deacetylase Inhibitors in HIV Latency Reversal. AIDS Res. Hum. Retroviruses 2022, 38, 615–621. [Google Scholar] [CrossRef]
- Wei, D.G.; Chiang, V.; Fyne, E.; Balakrishnan, M.; Barnes, T.; Graupe, M.; Hesselgesser, J.; Irrinki, A.; Murry, J.P.; Stepan, G.; et al. Histone deacetylase inhibitor romidepsin induces HIV expression in CD4 T cells from patients on suppressive antiretroviral therapy at concentrations achieved by clinical dosing. PLoS Pathog. 2014, 10, e1004071. [Google Scholar] [CrossRef] [PubMed]
- Banga, R.; Procopio, F.A.; Cavassini, M.; Perreau, M. In Vitro Reactivation of Replication-Competent and Infectious HIV-1 by Histone Deacetylase Inhibitors. J. Virol. 2016, 90, 1858–1871. [Google Scholar] [CrossRef] [PubMed]
- Shultz, M.; Fan, J.; Chen, C.; Cho, Y.S.; Davis, N.; Bickford, S.; Buteau, K.; Cao, X.; Holmqvist, M.; Hsu, M.; et al. The design, synthesis and structure-activity relationships of novel isoindoline-based histone deacetylase inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 4909–4912. [Google Scholar] [CrossRef] [PubMed]
- New Drugs/Drug News. Pharm. Ther. 2014, 39, 539–578.
- Younes, A.; Oki, Y.; Bociek, R.G.; Kuruvilla, J.; Fanale, M.; Neelapu, S.; Copeland, A.; Buglio, D.; Galal, A.; Besterman, J.; et al. Mocetinostat for relapsed classical Hodgkin’s lymphoma: An open-label, single-arm, phase 2 trial. Lancet Oncol. 2011, 12, 1222–1228. [Google Scholar] [CrossRef]
- Huber, K.; Doyon, G.; Plaks, J.; Fyne, E.; Mellors, J.W.; Sluis-Cremer, N. Inhibitors of histone deacetylases: Correlation between isoform specificity and reactivation of HIV type 1 (HIV-1) from latently infected cells. J. Biol. Chem. 2011, 286, 22211–22218. [Google Scholar] [CrossRef]
- Lu, W.; Yang, C.; Xu, X.; Chen, C.; Hou, X.; Fang, H.; Liu, S. A novel selective histone deacetylase I inhibitor CC-4a activates latent HIV-1 through NF-kappaB pathway. Life Sci. 2021, 267, 118427. [Google Scholar] [CrossRef]
- Blankstein, A.; Rubinstein, E.; Ezra, E.; Lokiec, F.; Caspi, I.; Horoszowski, H. Disc space infection and vertebral osteomyelitis as a complication of percutaneous lateral discectomy. Clin. Orthop. Relat. Res. 1987, 225, 234–237. [Google Scholar] [CrossRef]
- Friedman, J.; Cho, W.K.; Chu, C.K.; Keedy, K.S.; Archin, N.M.; Margolis, D.M.; Karn, J. Epigenetic silencing of HIV-1 by the histone H3 lysine 27 methyltransferase enhancer of Zeste 2. J. Virol. 2011, 85, 9078–9089. [Google Scholar] [CrossRef]
- Maina, E.K.; Adan, A.A.; Mureithi, H.; Muriuki, J.; Lwembe, R.M. A Review of Current Strategies Towards the Elimination of Latent HIV-1 and Subsequent HIV-1 Cure. Curr. HIV Res. 2021, 19, 14–26. [Google Scholar] [CrossRef]
- Kumar, A.; Darcis, G.; Van Lint, C.; Herbein, G. Epigenetic control of HIV-1 post integration latency: Implications for therapy. Clin. Epigenetics 2015, 7, 103. [Google Scholar] [CrossRef]
- Boehm, D.; Ott, M. Host Methyltransferases and Demethylases: Potential New Epigenetic Targets for HIV Cure Strategies and Beyond. AIDS Res. Hum. Retroviruses 2017, 33, S8–S22. [Google Scholar] [CrossRef]
- Bouchat, S.; Gatot, J.S.; Kabeya, K.; Cardona, C.; Colin, L.; Herbein, G.; De Wit, S.; Clumeck, N.; Lambotte, O.; Rouzioux, C.; et al. Histone methyltransferase inhibitors induce HIV-1 recovery in resting CD4(+) T cells from HIV-1-infected HAART-treated patients. AIDS 2012, 26, 1473–1482. [Google Scholar] [CrossRef]
- Margolis, D.M.; Archin, N.M.; Cohen, M.S.; Eron, J.J.; Ferrari, G.; Garcia, J.V.; Gay, C.L.; Goonetilleke, N.; Joseph, S.B.; Swanstrom, R.; et al. Curing HIV: Seeking to Target and Clear Persistent Infection. Cell 2020, 181, 189–206. [Google Scholar] [CrossRef]
- Wu, S.Y.; Chiang, C.M. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J. Biol. Chem. 2007, 282, 13141–13145. [Google Scholar] [CrossRef]
- Zaware, N.; Zhou, M.M. Bromodomain biology and drug discovery. Nat. Struct. Mol. Biol. 2019, 26, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.L.; Kim, C.; Zhou, M.M. The Functions of BET Proteins in Gene Transcription of Biology and Diseases. Front. Mol. Biosci. 2021, 8, 728777. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Lee, A.Y.; Lai, H.T.; Zhang, H.; Chiang, C.M. Phospho switch triggers Brd4 chromatin binding and activator recruitment for gene-specific targeting. Mol. Cell 2013, 49, 843–857. [Google Scholar] [CrossRef] [PubMed]
- Sims, R.J., 3rd; Belotserkovskaya, R.; Reinberg, D. Elongation by RNA polymerase II: The short and long of it. Genes Dev. 2004, 18, 2437–2468. [Google Scholar] [CrossRef]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Yik, J.H.; Chen, R.; He, N.; Jang, M.K.; Ozato, K.; Zhou, Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 2005, 19, 535–545. [Google Scholar] [CrossRef]
- Itzen, F.; Greifenberg, A.K.; Bosken, C.A.; Geyer, M. Brd4 activates P-TEFb for RNA polymerase II CTD phosphorylation. Nucleic Acids Res. 2014, 42, 7577–7590. [Google Scholar] [CrossRef]
- Bisgrove, D.A.; Mahmoudi, T.; Henklein, P.; Verdin, E. Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc. Natl. Acad. Sci. USA 2007, 104, 13690–13695. [Google Scholar] [CrossRef]
- Asamitsu, K.; Fujinaga, K.; Okamoto, T. HIV Tat/P-TEFb Interaction: A Potential Target for Novel Anti-HIV Therapies. Molecules 2018, 23, 933. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Guo, J.; Wu, Y.; Zhou, Q. The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing Brd4 inhibition of Tat-transactivation. Nucleic Acids Res. 2013, 41, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Gaiha, G.D.; John, S.P.; Pertel, T.; Chin, C.R.; Gao, G.; Qu, H.; Walker, B.D.; Elledge, S.J.; Brass, A.L. Reactivation of latent HIV-1 by inhibition of BRD4. Cell Rep. 2012, 2, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Boehm, D.; Calvanese, V.; Dar, R.D.; Xing, S.; Schroeder, S.; Martins, L.; Aull, K.; Li, P.C.; Planelles, V.; Bradner, J.E.; et al. BET bromodomain-targeting compounds reactivate HIV from latency via a Tat-independent mechanism. Cell Cycle 2013, 12, 452–462. [Google Scholar] [CrossRef]
- Boehm, D.; Conrad, R.J.; Ott, M. Bromodomain proteins in HIV infection. Viruses 2013, 5, 1571–1586. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Vakoc, C.R. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol. Cell 2014, 54, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Devaiah, B.N.; Gegonne, A.; Singer, D.S. Bromodomain 4: A cellular Swiss army knife. J. Leukoc. Biol. 2016, 100, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Salahong, T.; Schwartz, C.; Sungthong, R. Are BET Inhibitors yet Promising Latency-Reversing Agents for HIV-1 Reactivation in AIDS Therapy? Viruses 2021, 13, 1026. [Google Scholar] [CrossRef] [PubMed]
- Baldan, F.; Allegri, L.; Lazarevic, M.; Catia, M.; Milosevic, M.; Damante, G.; Milasin, J. Biological and molecular effects of bromodomain and extra-terminal (BET) inhibitors JQ1, IBET-151, and IBET-762 in OSCC cells. J. Oral Pathol. Med. 2019, 48, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Bauer, K.; Berger, D.; Zielinski, C.C.; Valent, P.; Grunt, T.W. Hitting two oncogenic machineries in cancer cells: Cooperative effects of the multi-kinase inhibitor ponatinib and the BET bromodomain blockers JQ1 or dBET1 on human carcinoma cells. Oncotarget 2018, 9, 26491–26506. [Google Scholar] [CrossRef] [PubMed]
- Bechter, O.; Schoffski, P. Make your best BET: The emerging role of BET inhibitor treatment in malignant tumors. Pharmacol. Ther. 2020, 208, 107479. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Yang, Y.; Yang, L.; Tian, J.; Zhang, F.; Zhou, J.; Zhang, H. 3-Hydroxyisoindolin-1-one derivates: Synthesis by palladium-catalyzed CH activation as BRD4 inhibitors against human acute myeloid leukemia (AML) cells. Bioorg. Chem. 2019, 86, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xu, L.; Mayakonda, A.; Huang, M.L.; Kanojia, D.; Tan, T.Z.; Dakle, P.; Lin, R.Y.; Ke, X.Y.; Said, J.W.; et al. Bromodomain and extraterminal proteins foster the core transcriptional regulatory programs and confer vulnerability in liposarcoma. Nat. Commun. 2019, 10, 1353. [Google Scholar] [CrossRef]
- Banerjee, C.; Archin, N.; Michaels, D.; Belkina, A.C.; Denis, G.V.; Bradner, J.; Sebastiani, P.; Margolis, D.M.; Montano, M. BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. J. Leukoc. Biol. 2012, 92, 1147–1154. [Google Scholar] [CrossRef]
- Conrad, R.J.; Fozouni, P.; Thomas, S.; Sy, H.; Zhang, Q.; Zhou, M.M.; Ott, M. The Short Isoform of BRD4 Promotes HIV-1 Latency by Engaging Repressive SWI/SNF Chromatin-Remodeling Complexes. Mol. Cell 2017, 67, 1001–1012. [Google Scholar] [CrossRef] [PubMed]
- Bullen, C.K.; Laird, G.M.; Durand, C.M.; Siliciano, J.D.; Siliciano, R.F. New ex vivo approaches distinguish effective and ineffective single agents for reversing HIV-1 latency in vivo. Nat. Med. 2014, 20, 425–429. [Google Scholar] [CrossRef]
- Li, G.; Zhang, Z.; Reszka-Blanco, N.; Li, F.; Chi, L.; Ma, J.; Jeffrey, J.; Cheng, L.; Su, L. Specific Activation In Vivo of HIV-1 by a Bromodomain Inhibitor from Monocytic Cells in Humanized Mice under Antiretroviral Therapy. J. Virol. 2019, 93, e00233-19. [Google Scholar] [CrossRef]
- Lu, P.; Qu, X.; Shen, Y.; Jiang, Z.; Wang, P.; Zeng, H.; Ji, H.; Deng, J.; Yang, X.; Li, X.; et al. The BET inhibitor OTX015 reactivates latent HIV-1 through P-TEFb. Sci. Rep. 2016, 6, 24100. [Google Scholar] [CrossRef]
- Liang, T.; Zhang, X.; Lai, F.; Lin, J.; Zhou, C.; Xu, X.; Tan, X.; Liu, S.; Li, L. A novel bromodomain inhibitor, CPI-203, serves as an HIV-1 latency-reversing agent by activating positive transcription elongation factor b. Biochem. Pharmacol. 2019, 164, 237–251. [Google Scholar] [CrossRef]
- Abner, E.; Stoszko, M.; Zeng, L.; Chen, H.C.; Izquierdo-Bouldstridge, A.; Konuma, T.; Zorita, E.; Fanunza, E.; Zhang, Q.; Mahmoudi, T.; et al. A New Quinoline BRD4 Inhibitor Targets a Distinct Latent HIV-1 Reservoir for Reactivation from Other “Shock” Drugs. J. Virol. 2018, 92, e02056-17. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Shen, Y.; Yang, H.; Wang, Y.; Jiang, Z.; Yang, X.; Zhong, Y.; Pan, H.; Xu, J.; Lu, H.; et al. BET inhibitors RVX-208 and PFI-1 reactivate HIV-1 from latency. Sci. Rep. 2017, 7, 16646. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Tyler, D.S.; Vappiani, J.; Caneque, T.; Lam, E.Y.N.; Ward, A.; Gilan, O.; Chan, Y.C.; Hienzsch, A.; Rutkowska, A.; Werner, T.; et al. Click chemistry enables preclinical evaluation of targeted epigenetic therapies. Science 2017, 356, 1397–1401. [Google Scholar] [CrossRef]
- Picaud, S.; Wells, C.; Felletar, I.; Brotherton, D.; Martin, S.; Savitsky, P.; Diez-Dacal, B.; Philpott, M.; Bountra, C.; Lingard, H.; et al. RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc. Natl. Acad. Sci. USA 2013, 110, 19754–19759. [Google Scholar] [CrossRef]
- Iwasaki, A.; Medzhitov, R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 2004, 5, 987–995. [Google Scholar] [CrossRef]
- Janeway, C.A., Jr.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef]
- Medzhitov, R.; Janeway, C.A., Jr. Innate immunity: The virtues of a nonclonal system of recognition. Cell 1997, 91, 295–298. [Google Scholar] [CrossRef]
- Medzhitov, R.; Preston-Hurlburt, P.; Janeway, C.A., Jr. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 1997, 388, 394–397. [Google Scholar] [CrossRef]
- Martinsen, J.T.; Gunst, J.D.; Hojen, J.F.; Tolstrup, M.; Sogaard, O.S. The Use of Toll-Like Receptor Agonists in HIV-1 Cure Strategies. Front. Immunol. 2020, 11, 1112. [Google Scholar] [CrossRef] [PubMed]
- Macedo, A.B.; Novis, C.L.; Bosque, A. Targeting Cellular and Tissue HIV Reservoirs With Toll-Like Receptor Agonists. Front. Immunol. 2019, 10, 2450. [Google Scholar] [CrossRef]
- Liu, G.; Zhao, Y. Toll-like receptors and immune regulation: Their direct and indirect modulation on regulatory CD4+ CD25+ T cells. Immunology 2007, 122, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Saxena, M.; Sabado, R.L.; La Mar, M.; Mohri, H.; Salazar, A.M.; Dong, H.; Correa Da Rosa, J.; Markowitz, M.; Bhardwaj, N.; Miller, E. Poly-ICLC, a TLR3 Agonist, Induces Transient Innate Immune Responses in Patients With Treated HIV-Infection: A Randomized Double-Blinded Placebo Controlled Trial. Front. Immunol. 2019, 10, 725. [Google Scholar] [CrossRef]
- Borducchi, E.N.; Cabral, C.; Stephenson, K.E.; Liu, J.; Abbink, P.; Ng’ang’a, D.; Nkolola, J.P.; Brinkman, A.L.; Peter, L.; Lee, B.C.; et al. Ad26/MVA therapeutic vaccination with TLR7 stimulation in SIV-infected rhesus monkeys. Nature 2016, 540, 284–287. [Google Scholar] [CrossRef]
- Borducchi, E.N.; Liu, J.; Nkolola, J.P.; Cadena, A.M.; Yu, W.H.; Fischinger, S.; Broge, T.; Abbink, P.; Mercado, N.B.; Chandrashekar, A.; et al. Antibody and TLR7 agonist delay viral rebound in SHIV-infected monkeys. Nature 2018, 563, 360–364. [Google Scholar] [CrossRef]
- Lim, S.Y.; Osuna, C.E.; Hraber, P.T.; Hesselgesser, J.; Gerold, J.M.; Barnes, T.L.; Sanisetty, S.; Seaman, M.S.; Lewis, M.G.; Geleziunas, R.; et al. TLR7 agonists induce transient viremia and reduce the viral reservoir in SIV-infected rhesus macaques on antiretroviral therapy. Sci. Transl. Med. 2018, 10, eaao4521. [Google Scholar] [CrossRef]
- Del Prete, G.Q.; Alvord, W.G.; Li, Y.; Deleage, C.; Nag, M.; Oswald, K.; Thomas, J.A.; Pyle, C.; Bosche, W.J.; Coalter, V.; et al. TLR7 agonist administration to SIV-infected macaques receiving early initiated cART does not induce plasma viremia. JCI Insight 2019, 4, e127717. [Google Scholar] [CrossRef] [PubMed]
- Bekerman, E.; Hesselgesser, J.; Carr, B.; Nagel, M.; Hung, M.; Wang, A.; Stapleton, L.; von Gegerfelt, A.; Elyard, H.A.; Lifson, J.D.; et al. PD-1 Blockade and TLR7 Activation Lack Therapeutic Benefit in Chronic Simian Immunodeficiency Virus-Infected Macaques on Antiretroviral Therapy. Antimicrob. Agents Chemother. 2019, 63, e01163-19. [Google Scholar] [CrossRef] [PubMed]
- Griffin, G.E.; Leung, K.; Folks, T.M.; Kunkel, S.; Nabel, G.J. Activation of HIV gene expression during monocyte differentiation by induction of NF-kappa B. Nature 1989, 339, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Dandekar, S. Targeting NF-kappaB signaling with protein kinase C agonists as an emerging strategy for combating HIV latency. AIDS Res. Hum. Retroviruses 2015, 31, 4–12. [Google Scholar] [CrossRef]
- McKernan, L.N.; Momjian, D.; Kulkosky, J. Protein Kinase C: One Pathway towards the Eradication of Latent HIV-1 Reservoirs. Adv. Virol. 2012, 2012, 805347. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.H.; Grobler, J.A. Protein kinase C and phospholipase C: Bilayer interactions and regulation. Curr. Opin. Struct. Biol. 1997, 7, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Goel, G.; Makkar, H.P.; Francis, G.; Becker, K. Phorbol esters: Structure, biological activity, and toxicity in animals. Int. J. Toxicol. 2007, 26, 279–288. [Google Scholar] [CrossRef]
- Rullas, J.; Bermejo, M.; Garcia-Perez, J.; Beltan, M.; Gonzalez, N.; Hezareh, M.; Brown, S.J.; Alcami, J. Prostratin induces HIV activation and downregulates HIV receptors in peripheral blood lymphocytes. Antivir. Ther. 2004, 9, 545–554. [Google Scholar] [CrossRef]
- Philip, P.A.; Rea, D.; Thavasu, P.; Carmichael, J.; Stuart, N.S.; Rockett, H.; Talbot, D.C.; Ganesan, T.; Pettit, G.R.; Balkwill, F.; et al. Phase I study of bryostatin 1: Assessment of interleukin 6 and tumor necrosis factor alpha induction in vivo. The Cancer Research Campaign Phase I Committee. J. Natl. Cancer Inst. 1993, 85, 1812–1818. [Google Scholar] [CrossRef]
- Jayson, G.C.; Crowther, D.; Prendiville, J.; McGown, A.T.; Scheid, C.; Stern, P.; Young, R.; Brenchley, P.; Chang, J.; Owens, S.; et al. A phase I trial of bryostatin 1 in patients with advanced malignancy using a 24 hour intravenous infusion. Br. J. Cancer 1995, 72, 461–468. [Google Scholar] [CrossRef]
- Mehla, R.; Bivalkar-Mehla, S.; Zhang, R.; Handy, I.; Albrecht, H.; Giri, S.; Nagarkatti, P.; Nagarkatti, M.; Chauhan, A. Bryostatin modulates latent HIV-1 infection via PKC and AMPK signaling but inhibits acute infection in a receptor independent manner. PLoS ONE 2010, 5, e11160. [Google Scholar] [CrossRef]
- Curreli, F.; Ahmed, S.; Victor, S.M.B.; Debnath, A.K. Identification of Combinations of Protein Kinase C Activators and Histone Deacetylase Inhibitors That Potently Reactivate Latent HIV. Viruses 2020, 12, 609. [Google Scholar] [CrossRef]
- Steinberg, S.F. Structural basis of protein kinase C isoform function. Physiol. Rev. 2008, 88, 1341–1378. [Google Scholar] [CrossRef] [PubMed]
- von Burstin, V.A.; Xiao, L.; Kazanietz, M.G. Bryostatin 1 inhibits phorbol ester-induced apoptosis in prostate cancer cells by differentially modulating protein kinase C (PKC) delta translocation and preventing PKCdelta-mediated release of tumor necrosis factor-alpha. Mol. Pharmacol. 2010, 78, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Suman Ranjan Das, S.J. Biology of the HIV Nef protein. Indian J. Med. Res. 2005, 121, 315–332. [Google Scholar]
- Wolf, D.; Witte, V.; Laffert, B.; Blume, K.; Stromer, E.; Trapp, S.; d’Aloja, P.; Schurmann, A.; Baur, A.S. HIV-1 Nef associated PAK and PI3-kinases stimulate Akt-independent Bad-phosphorylation to induce anti-apoptotic signals. Nat. Med. 2001, 7, 1217–1224. [Google Scholar] [CrossRef]
- Deng, X.; Ruvolo, P.; Carr, B.; May, W.S., Jr. Survival function of ERK1/2 as IL-3-activated, staurosporine-resistant Bcl2 kinases. Proc. Natl. Acad. Sci. USA 2000, 97, 1578–1583. [Google Scholar] [CrossRef] [PubMed]
- French, A.J.; Natesampillai, S.; Krogman, A.; Correia, C.; Peterson, K.L.; Alto, A.; Chandrasekar, A.P.; Misra, A.; Li, Y.; Kaufmann, S.H.; et al. Reactivating latent HIV with PKC agonists induces resistance to apoptosis and is associated with phosphorylation and activation of BCL2. PLoS Pathog. 2020, 16, e1008906. [Google Scholar] [CrossRef]
- Morgan, R.J., Jr.; Leong, L.; Chow, W.; Gandara, D.; Frankel, P.; Garcia, A.; Lenz, H.J.; Doroshow, J.H. Phase II trial of bryostatin-1 in combination with cisplatin in patients with recurrent or persistent epithelial ovarian cancer: A California cancer consortium study. Investig. New Drugs 2012, 30, 723–728. [Google Scholar] [CrossRef]
- Jiang, G.; Maverakis, E.; Cheng, M.Y.; Elsheikh, M.M.; Deleage, C.; Mendez-Lagares, G.; Shimoda, M.; Yukl, S.A.; Hartigan-O’Connor, D.J.; Thompson, G.R., 3rd; et al. Disruption of latent HIV in vivo during the clearance of actinic keratosis by ingenol mebutate. JCI Insight 2019, 4, e126027. [Google Scholar] [CrossRef]
- Fujiwara, M.; Okamoto, M.; Ijichi, K.; Tokuhisa, K.; Hanasaki, Y.; Katsuura, K.; Uemura, D.; Shigeta, S.; Konno, K.; Yokota, T.; et al. Upregulation of HIV-1 replication in chronically infected cells by ingenol derivatives. Arch. Virol. 1998, 143, 2003–2010. [Google Scholar] [CrossRef]
- Pandelo Jose, D.; Bartholomeeusen, K.; da Cunha, R.D.; Abreu, C.M.; Glinski, J.; da Costa, T.B.; Bacchi Rabay, A.F.; Pianowski Filho, L.F.; Dudycz, L.W.; Ranga, U.; et al. Reactivation of latent HIV-1 by new semi-synthetic ingenol esters. Virology 2014, 462–463, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Abreu, C.M.; Price, S.L.; Shirk, E.N.; Cunha, R.D.; Pianowski, L.F.; Clements, J.E.; Tanuri, A.; Gama, L. Dual role of novel ingenol derivatives from Euphorbia tirucalli in HIV replication: Inhibition of de novo infection and activation of viral LTR. PLoS ONE 2014, 9, e97257. [Google Scholar] [CrossRef]
- Warrilow, D.; Gardner, J.; Darnell, G.A.; Suhrbier, A.; Harrich, D. HIV type 1 inhibition by protein kinase C modulatory compounds. AIDS Res. Hum. Retroviruses 2006, 22, 854–864. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Mendes, E.A.; Kaiser, P.; Wong, D.P.; Tang, Y.; Cai, I.; Fenton, A.; Melcher, G.P.; Hildreth, J.E.; Thompson, G.R.; et al. Synergistic Reactivation of Latent HIV Expression by Ingenol-3-Angelate, PEP005, Targeted NF-kB Signaling in Combination with JQ1 Induced p-TEFb Activation. PLoS Pathog. 2015, 11, e1005066. [Google Scholar] [CrossRef] [PubMed]
- Gama, L.; Abreu, C.M.; Shirk, E.N.; Price, S.L.; Li, M.; Laird, G.M.; Pate, K.A.; Wietgrefe, S.W.; O’Connor, S.L.; Pianowski, L.; et al. Reactivation of simian immunodeficiency virus reservoirs in the brain of virally suppressed macaques. AIDS 2017, 31, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Okoye, A.A.; Fromentin, R.; Takata, H.; Brehm, J.H.; Fukazawa, Y.; Randall, B.; Pardons, M.; Tai, V.; Tang, J.; Smedley, J.; et al. The ingenol-based protein kinase C agonist GSK445A is a potent inducer of HIV and SIV RNA transcription. PLoS Pathog. 2022, 18, e1010245. [Google Scholar] [CrossRef]
- Asada, Y.; Sukemori, A.; Watanabe, T.; Malla, K.J.; Yoshikawa, T.; Li, W.; Koike, K.; Chen, C.H.; Akiyama, T.; Qian, K.; et al. Stelleralides A-C, novel potent anti-HIV daphnane-type diterpenoids from Stellera chamaejasme L. Org. Lett. 2011, 13, 2904–2907. [Google Scholar] [CrossRef]
- Huang, L.; Ho, P.; Yu, J.; Zhu, L.; Lee, K.H.; Chen, C.H. Picomolar dichotomous activity of gnidimacrin against HIV-1. PLoS ONE 2011, 6, e26677. [Google Scholar] [CrossRef]
- Asada, Y.; Sukemori, A.; Watanabe, T.; Malla, K.J.; Yoshikawa, T.; Li, W.; Kuang, X.; Koike, K.; Chen, C.H.; Akiyama, T.; et al. Isolation, structure determination, and anti-HIV evaluation of tigliane-type diterpenes and biflavonoid from Stellera chamaejasme. J. Nat. Prod. 2013, 76, 852–857. [Google Scholar] [CrossRef]
- Lai, W.; Huang, L.; Zhu, L.; Ferrari, G.; Chan, C.; Li, W.; Lee, K.H.; Chen, C.H. Gnidimacrin, a Potent Anti-HIV Diterpene, Can Eliminate Latent HIV-1 Ex Vivo by Activation of Protein Kinase C beta. J. Med. Chem. 2015, 58, 8638–8646. [Google Scholar] [CrossRef] [PubMed]
- Xing, S.; Bullen, C.K.; Shroff, N.S.; Shan, L.; Yang, H.C.; Manucci, J.L.; Bhat, S.; Zhang, H.; Margolick, J.B.; Quinn, T.C.; et al. Disulfiram reactivates latent HIV-1 in a Bcl-2-transduced primary CD4+ T cell model without inducing global T cell activation. J. Virol. 2011, 85, 6060–6064. [Google Scholar] [CrossRef] [PubMed]
- Doyon, G.; Zerbato, J.; Mellors, J.W.; Sluis-Cremer, N. Disulfiram reactivates latent HIV-1 expression through depletion of the phosphatase and tensin homolog. AIDS 2013, 27, F7–F11. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.H.; McMahon, J.H.; Chang, C.C.; Lee, S.A.; Hartogensis, W.; Bumpus, N.; Savic, R.; Roney, J.; Hoh, R.; Solomon, A.; et al. Short-term administration of disulfiram for reversal of latent HIV infection: A phase 2 dose-escalation study. Lancet HIV 2015, 2, e520–e529. [Google Scholar] [CrossRef]
- Spivak, A.M.; Andrade, A.; Eisele, E.; Hoh, R.; Bacchetti, P.; Bumpus, N.N.; Emad, F.; Buckheit, R., 3rd; McCance-Katz, E.F.; Lai, J.; et al. A pilot study assessing the safety and latency-reversing activity of disulfiram in HIV-1-infected adults on antiretroviral therapy. Clin. Infect. Dis. 2014, 58, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Laird, G.M.; Bullen, C.K.; Rosenbloom, D.I.; Martin, A.R.; Hill, A.L.; Durand, C.M.; Siliciano, J.D.; Siliciano, R.F. Ex vivo analysis identifies effective HIV-1 latency-reversing drug combinations. J. Clin. Investig. 2015, 125, 1901–1912. [Google Scholar] [CrossRef] [PubMed]
- Pache, L.; Dutra, M.S.; Spivak, A.M.; Marlett, J.M.; Murry, J.P.; Hwang, Y.; Maestre, A.M.; Manganaro, L.; Vamos, M.; Teriete, P.; et al. BIRC2/cIAP1 Is a Negative Regulator of HIV-1 Transcription and Can Be Targeted by Smac Mimetics to Promote Reversal of Viral Latency. Cell Host Microbe 2015, 18, 345–353. [Google Scholar] [CrossRef]
- Sun, S.C. The noncanonical NF-kappaB pathway. Immunol. Rev. 2012, 246, 125–140. [Google Scholar] [CrossRef]
- Nixon, C.C.; Mavigner, M.; Sampey, G.C.; Brooks, A.D.; Spagnuolo, R.A.; Irlbeck, D.M.; Mattingly, C.; Ho, P.T.; Schoof, N.; Cammon, C.G.; et al. Systemic HIV and SIV latency reversal via non-canonical NF-kappaB signalling in vivo. Nature 2020, 578, 160–165. [Google Scholar] [CrossRef]
- Pache, L.; Marsden, M.D.; Teriete, P.; Portillo, A.J.; Heimann, D.; Kim, J.T.; Soliman, M.S.A.; Dimapasoc, M.; Carmona, C.; Celeridad, M.; et al. Pharmacological Activation of Non-canonical NF-kappaB Signaling Activates Latent HIV-1 Reservoirs In Vivo. Cell Rep. Med. 2020, 1, 100037. [Google Scholar] [CrossRef]
- Berro, R.; de la Fuente, C.; Klase, Z.; Kehn, K.; Parvin, L.; Pumfery, A.; Agbottah, E.; Vertes, A.; Nekhai, S.; Kashanchi, F. Identifying the membrane proteome of HIV-1 latently infected cells. J. Biol. Chem. 2007, 282, 8207–8218. [Google Scholar] [CrossRef] [PubMed]
- Eckelman, B.P.; Salvesen, G.S. The human anti-apoptotic proteins cIAP1 and cIAP2 bind but do not inhibit caspases. J. Biol. Chem. 2006, 281, 3254–3260. [Google Scholar] [CrossRef] [PubMed]
- Bobardt, M.; Kuo, J.; Chatterji, U.; Chanda, S.; Little, S.J.; Wiedemann, N.; Vuagniaux, G.; Gallay, P.A. The inhibitor apoptosis protein antagonist Debio 1143 Is an attractive HIV-1 latency reversal candidate. PLoS ONE 2019, 14, e0211746. [Google Scholar] [CrossRef] [PubMed]
- Dashti, A.; Sukkestad, S.; Horner, A.M.; Neja, M.; Siddiqi, Z.; Waller, C.; Goldy, J.; Monroe, D.; Lin, A.; Schoof, N.; et al. AZD5582 plus SIV-specific antibodies reduce lymph node viral reservoirs in antiretroviral therapy-suppressed macaques. Nat. Med. 2023, 29, 2535–2546. [Google Scholar] [CrossRef]
- Lewin, S.R.; Attoye, T.; Bansbach, C.; Doehle, B.; Dube, K.; Dybul, M.; SenGupta, D.; Jiang, A.; Johnston, R.; Lamplough, R.; et al. Multi-stakeholder consensus on a target product profile for an HIV cure. Lancet HIV 2021, 8, e42–e50. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Bonet, M.; Clemente, M.I.; Serramia, M.J.; Munoz, E.; Moreno, S.; Munoz-Fernandez, M.A. Synergistic Activation of Latent HIV-1 Expression by Novel Histone Deacetylase Inhibitors and Bryostatin-1. Sci. Rep. 2015, 5, 16445. [Google Scholar] [CrossRef] [PubMed]
- Reuse, S.; Calao, M.; Kabeya, K.; Guiguen, A.; Gatot, J.S.; Quivy, V.; Vanhulle, C.; Lamine, A.; Vaira, D.; Demonte, D.; et al. Synergistic activation of HIV-1 expression by deacetylase inhibitors and prostratin: Implications for treatment of latent infection. PLoS ONE 2009, 4, e6093. [Google Scholar] [CrossRef]
- Pardons, M.; Fromentin, R.; Pagliuzza, A.; Routy, J.P.; Chomont, N. Latency-Reversing Agents Induce Differential Responses in Distinct Memory CD4 T Cell Subsets in Individuals on Antiretroviral Therapy. Cell Rep. 2019, 29, 2783–2795. [Google Scholar] [CrossRef]
- Darcis, G.; Kula, A.; Bouchat, S.; Fujinaga, K.; Corazza, F.; Ait-Ammar, A.; Delacourt, N.; Melard, A.; Kabeya, K.; Vanhulle, C.; et al. An In-Depth Comparison of Latency-Reversing Agent Combinations in Various In Vitro and Ex Vivo HIV-1 Latency Models Identified Bryostatin-1+JQ1 and Ingenol-B+JQ1 to Potently Reactivate Viral Gene Expression. PLoS Pathog. 2015, 11, e1005063. [Google Scholar] [CrossRef]
- Matsuda, K.; Kobayakawa, T.; Kariya, R.; Tsuchiya, K.; Ryu, S.; Tsuji, K.; Ishii, T.; Gatanaga, H.; Yoshimura, K.; Okada, S.; et al. A Therapeutic Strategy to Combat HIV-1 Latently Infected Cells With a Combination of Latency-Reversing Agents Containing DAG-Lactone PKC Activators. Front. Microbiol. 2021, 12, 636276. [Google Scholar] [CrossRef]
- Rodari, A.; Darcis, G.; Van Lint, C.M. The Current Status of Latency Reversing Agents for HIV-1 Remission. Annu. Rev. Virol. 2021, 8, 491–514. [Google Scholar] [CrossRef]
- Huang, H.; Liu, S.; Jean, M.; Simpson, S.; Huang, H.; Merkley, M.; Hayashi, T.; Kong, W.; Rodriguez-Sanchez, I.; Zhang, X.; et al. A Novel Bromodomain Inhibitor Reverses HIV-1 Latency through Specific Binding with BRD4 to Promote Tat and P-TEFb Association. Front. Microbiol. 2017, 8, 1035. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.X.; Lin, J.; Liang, T.Z.; Duan, H.; Tan, X.H.; Xi, B.M.; Li, L.; Liu, S.W. The BET bromodomain inhibitor apabetalone induces apoptosis of latent HIV-1 reservoir cells following viral reactivation. Acta Pharmacol. Sin. 2019, 40, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Washizaki, A.; Murata, M.; Seki, Y.; Kikumori, M.; Tang, Y.; Tan, W.; Wardani, N.P.; Irie, K.; Akari, H. The Novel PKC Activator 10-Methyl-Aplog-1 Combined with JQ1 Induced Strong and Synergistic HIV Reactivation with Tolerable Global T Cell Activation. Viruses 2021, 13, 2037. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, Y.; Kudo, K.; Zeligs, K.P.; Anderson, D.; Hernandez, L.; Ning, F.; Cole, C.B.; Fergusson, M.; Kedei, N.; Lyons, J.; et al. SMAC Mimetics Synergistically Cooperate with HDAC Inhibitors Enhancing TNF-alpha Autocrine Signaling. Cancers 2023, 15, 1315. [Google Scholar] [CrossRef] [PubMed]
- Falcinelli, S.D.; Peterson, J.J.; Turner, A.W.; Irlbeck, D.; Read, J.; Raines, S.L.; James, K.S.; Sutton, C.; Sanchez, A.; Emery, A.; et al. Combined noncanonical NF-kappaB agonism and targeted BET bromodomain inhibition reverse HIV latency ex vivo. J. Clin. Investig. 2022, 132, e157281. [Google Scholar] [CrossRef]
- Hattori, S.I.; Matsuda, K.; Tsuchiya, K.; Gatanaga, H.; Oka, S.; Yoshimura, K.; Mitsuya, H.; Maeda, K. Combination of a Latency-Reversing Agent With a Smac Mimetic Minimizes Secondary HIV-1 Infection in vitro. Front. Microbiol. 2018, 9, 2022. [Google Scholar] [CrossRef] [PubMed]
- Molyer, B.; Kumar, A.; Angel, J.B. SMAC Mimetics as Therapeutic Agents in HIV Infection. Front. Immunol. 2021, 12, 780400. [Google Scholar] [CrossRef]
- Howard, J.N.; Bosque, A. IL-15 and N-803 for HIV Cure Approaches. Viruses 2023, 15, 1912. [Google Scholar] [CrossRef]
- Miller, J.S.; Davis, Z.B.; Helgeson, E.; Reilly, C.; Thorkelson, A.; Anderson, J.; Lima, N.S.; Jorstad, S.; Hart, G.T.; Lee, J.H.; et al. Safety and virologic impact of the IL-15 superagonist N-803 in people living with HIV: A phase 1 trial. Nat. Med. 2022, 28, 392–400. [Google Scholar] [CrossRef]
- Copertino, D.C., Jr.; Holmberg, C.S.; Weiler, J.; Ward, A.R.; Howard, J.N.; Levinger, C.; Pang, A.P.; Corley, M.J.; Dundar, F.; Zumbo, P.; et al. The latency-reversing agent HODHBt synergizes with IL-15 to enhance cytotoxic function of HIV-specific T cells. JCI Insight 2023, 8, e169028. [Google Scholar] [CrossRef] [PubMed]
- Bosque, A.; Nilson, K.A.; Macedo, A.B.; Spivak, A.M.; Archin, N.M.; Van Wagoner, R.M.; Martins, L.J.; Novis, C.L.; Szaniawski, M.A.; Ireland, C.M.; et al. Benzotriazoles Reactivate Latent HIV-1 through Inactivation of STAT5 SUMOylation. Cell Rep. 2017, 18, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Macedo, A.B.; Levinger, C.; Nguyen, B.N.; Richard, J.; Gupta, M.; Cruz, C.R.Y.; Finzi, A.; Chiappinelli, K.B.; Crandall, K.A.; Bosque, A. The HIV Latency Reversal Agent HODHBt Enhances NK Cell Effector and Memory-Like Functions by Increasing Interleukin-15-Mediated STAT Activation. J. Virol. 2022, 96, e0037222. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, E.S.; Macedo, A.B.; Resop, R.S.; Howard, J.N.; Nell, R.; Sarabia, I.; Newman, D.; Ren, Y.; Jones, R.B.; Planelles, V.; et al. Structure-Activity Relationship Analysis of Benzotriazine Analogues as HIV-1 Latency-Reversing Agents. Antimicrob. Agents Chemother. 2020, 64, e00888-20. [Google Scholar] [CrossRef]
- Soliman, S.H.A.; Cisneros, W.J.; Iwanaszko, M.; Aoi, Y.; Ganesan, S.; Walter, M.; Zeidner, J.M.; Mishra, R.K.; Kim, E.Y.; Wolinsky, S.M.; et al. Enhancing HIV-1 latency reversal through regulating the elongating RNA Pol II pause-release by a small-molecule disruptor of PAF1C. Sci. Adv. 2023, 9, eadf2468. [Google Scholar] [CrossRef] [PubMed]
- Clutton, G.T.; Jones, R.B. Diverse Impacts of HIV Latency-Reversing Agents on CD8+ T-Cell Function: Implications for HIV Cure. Front. Immunol. 2018, 9, 1452. [Google Scholar] [CrossRef]
- Kim, Y.; Anderson, J.L.; Lewin, S.R. Getting the “Kill” into “Shock and Kill”: Strategies to Eliminate Latent HIV. Cell Host Microbe 2018, 23, 14–26. [Google Scholar] [CrossRef]
- Ward, A.R.; Mota, T.M.; Jones, R.B. Immunological approaches to HIV cure. Semin. Immunol. 2021, 51, 101412. [Google Scholar] [CrossRef]
- Gupta, R.K.; Abdul-Jawad, S.; McCoy, L.E.; Mok, H.P.; Peppa, D.; Salgado, M.; Martinez-Picado, J.; Nijhuis, M.; Wensing, A.M.J.; Lee, H.; et al. HIV-1 remission following CCR5Delta32/Delta32 haematopoietic stem-cell transplantation. Nature 2019, 568, 244–248. [Google Scholar] [CrossRef]
- Hutter, G.; Nowak, D.; Mossner, M.; Ganepola, S.; Mussig, A.; Allers, K.; Schneider, T.; Hofmann, J.; Kucherer, C.; Blau, O.; et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N. Engl. J. Med. 2009, 360, 692–698. [Google Scholar] [CrossRef]
- Wang, C.X.; Cannon, P.M. The clinical applications of genome editing in HIV. Blood 2016, 127, 2546–2552. [Google Scholar] [CrossRef] [PubMed]
- Tebas, P.; Stein, D.; Tang, W.W.; Frank, I.; Wang, S.Q.; Lee, G.; Spratt, S.K.; Surosky, R.T.; Giedlin, M.A.; Nichol, G.; et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med. 2014, 370, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Tebas, P.; Jadlowsky, J.K.; Shaw, P.A.; Tian, L.; Esparza, E.; Brennan, A.L.; Kim, S.; Naing, S.Y.; Richardson, M.W.; Vogel, A.N.; et al. CCR5-edited CD4+ T cells augment HIV-specific immunity to enable post-rebound control of HIV replication. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Zeidan, J.; Sharma, A.A.; Lee, G.; Raad, A.; Fromentin, R.; Fourati, S.; Ghneim, K.; Sanchez, G.P.; Benne, C.; Canderan, G.; et al. Infusion of CCR5 Gene-Edited T Cells Allows Immune Reconstitution, HIV Reservoir Decay, and Long-Term Virological Control. bioRxiv 2021. [Google Scholar] [CrossRef]
- Ellwanger, J.H.; Kulmann-Leal, B.; Kaminski, V.L.; Rodrigues, A.G.; Bragatte, M.A.S.; Chies, J.A.B. Beyond HIV infection: Neglected and varied impacts of CCR5 and CCR5Delta32 on viral diseases. Virus Res. 2020, 286, 198040. [Google Scholar] [CrossRef]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef]
- Mitsuyasu, R.T.; Anton, P.A.; Deeks, S.G.; Scadden, D.T.; Connick, E.; Downs, M.T.; Bakker, A.; Roberts, M.R.; June, C.H.; Jalali, S.; et al. Prolonged survival and tissue trafficking following adoptive transfer of CD4zeta gene-modified autologous CD4(+) and CD8(+) T cells in human immunodeficiency virus-infected subjects. Blood 2000, 96, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Seif, M.; Einsele, H.; Loffler, J. CAR T Cells Beyond Cancer: Hope for Immunomodulatory Therapy of Infectious Diseases. Front. Immunol. 2019, 10, 2711. [Google Scholar] [CrossRef]
- Chmielewski, M.; Hombach, A.A.; Abken, H. Of CARs and TRUCKs: Chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol. Rev. 2014, 257, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Scholler, J.; Brady, T.L.; Binder-Scholl, G.; Hwang, W.T.; Plesa, G.; Hege, K.M.; Vogel, A.N.; Kalos, M.; Riley, J.L.; Deeks, S.G.; et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci. Transl. Med. 2012, 4, 132ra53. [Google Scholar] [CrossRef]
- Carvalho, T. First two patients receive CAR T cell therapy for HIV. Nat. Med. 2023, 29, 1290–1291. [Google Scholar] [CrossRef]
- Campos-Gonzalez, G.; Martinez-Picado, J.; Velasco-Hernandez, T.; Salgado, M. Opportunities for CAR-T Cell Immunotherapy in HIV Cure. Viruses 2023, 15, 789. [Google Scholar] [CrossRef] [PubMed]
- Nordstrom, J.L.; Ferrari, G.; Margolis, D.M. Bispecific antibody-derived molecules to target persistent HIV infection. J. Virus Erad. 2022, 8, 100083. [Google Scholar] [CrossRef] [PubMed]
- Evans, V.A.; van der Sluis, R.M.; Solomon, A.; Dantanarayana, A.; McNeil, C.; Garsia, R.; Palmer, S.; Fromentin, R.; Chomont, N.; Sekaly, R.P.; et al. Programmed cell death-1 contributes to the establishment and maintenance of HIV-1 latency. AIDS 2018, 32, 1491–1497. [Google Scholar] [CrossRef]
- Uldrick, T.S.; Adams, S.V.; Fromentin, R.; Roche, M.; Fling, S.P.; Goncalves, P.H.; Lurain, K.; Ramaswami, R.; Wang, C.J.; Gorelick, R.J.; et al. Pembrolizumab induces HIV latency reversal in people living with HIV and cancer on antiretroviral therapy. Sci. Transl. Med. 2022, 14, eabl3836. [Google Scholar] [CrossRef]
- Guihot, A.; Marcelin, A.G.; Massiani, M.A.; Samri, A.; Soulie, C.; Autran, B.; Spano, J.P. Drastic decrease of the HIV reservoir in a patient treated with nivolumab for lung cancer. Ann. Oncol. 2018, 29, 517–518. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, T.A.; Rajdev, L.; Rhodes, A.; Dantanarayana, A.; Tennakoon, S.; Chea, S.; Spelman, T.; Lensing, S.; Rutishauser, R.; Bakkour, S.; et al. Impact of Anti-PD-1 and Anti-CTLA-4 on the Human Immunodeficiency Virus (HIV) Reservoir in People Living With HIV With Cancer on Antiretroviral Therapy: The AIDS Malignancy Consortium 095 Study. Clin. Infect. Dis. 2021, 73, e1973–e1981. [Google Scholar] [CrossRef]
- Rust, B.J.; Kean, L.S.; Colonna, L.; Brandenstein, K.E.; Poole, N.H.; Obenza, W.; Enstrom, M.R.; Maldini, C.R.; Ellis, G.I.; Fennessey, C.M.; et al. Robust expansion of HIV CAR T cells following antigen boosting in ART-suppressed nonhuman primates. Blood 2020, 136, 1722–1734. [Google Scholar] [CrossRef]
- Wykes, M.N.; Lewin, S.R. Immune checkpoint blockade in infectious diseases. Nat. Rev. Immunol. 2018, 18, 91–104. [Google Scholar] [CrossRef]
- Kulpa, D.A.; Talla, A.; Brehm, J.H.; Ribeiro, S.P.; Yuan, S.; Bebin-Blackwell, A.G.; Miller, M.; Barnard, R.; Deeks, S.G.; Hazuda, D.; et al. Differentiation into an Effector Memory Phenotype Potentiates HIV-1 Latency Reversal in CD4(+) T Cells. J. Virol. 2019, 93, e00969-19. [Google Scholar] [CrossRef] [PubMed]
- Samer, S.; Thomas, Y.; Arainga, M.; Carter, C.; Shirreff, L.M.; Arif, M.S.; Avita, J.M.; Frank, I.; McRaven, M.D.; Thuruthiyil, C.T.; et al. Blockade of TGF-beta signaling reactivates HIV-1/SIV reservoirs and immune responses in vivo. JCI Insight 2022, 7, e162290. [Google Scholar] [CrossRef]
- Kime, J.; Bose, D.; Arainga, M.; Haque, M.R.; Fennessey, C.M.; Caddell, R.A.; Thomas, Y.; Ferrell, D.E.; Ali, S.; Grody, E.; et al. TGF-β blockade drives a transitional effector phenotype in T cells reversing SIV latency and decreasing SIV reservoirs in vivo. bioRxiv 2023. [Google Scholar] [CrossRef]
- Malim, M.H.; Emerman, M. HIV-1 accessory proteins--ensuring viral survival in a hostile environment. Cell Host Microbe 2008, 3, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Diehl, N.; Schaal, H. Make yourself at home: Viral hijacking of the PI3K/Akt signaling pathway. Viruses 2013, 5, 3192–3212. [Google Scholar] [CrossRef] [PubMed]
- Juarez-Salcedo, L.M.; Desai, V.; Dalia, S. Venetoclax: Evidence to date and clinical potential. Drugs Context 2019, 8, 212574. [Google Scholar] [CrossRef]
- de Vos, S.; Leonard, J.P.; Friedberg, J.W.; Zain, J.; Dunleavy, K.; Humerickhouse, R.; Hayslip, J.; Pesko, J.; Wilson, W.H. Safety and efficacy of navitoclax, a BCL-2 and BCL-X(L) inhibitor, in patients with relapsed or refractory lymphoid malignancies: Results from a phase 2a study. Leuk. Lymphoma 2021, 62, 810–818. [Google Scholar] [CrossRef]
- Marconi, V.C.; Moser, C.; Gavegnano, C.; Deeks, S.G.; Lederman, M.M.; Overton, E.T.; Tsibris, A.; Hunt, P.W.; Kantor, A.; Sekaly, R.P.; et al. Randomized Trial of Ruxolitinib in Antiretroviral-Treated Adults With Human Immunodeficiency Virus. Clin. Infect. Dis. 2022, 74, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Cummins, N.W.; Sainski, A.M.; Dai, H.; Natesampillai, S.; Pang, Y.P.; Bren, G.D.; de Araujo Correia, M.C.M.; Sampath, R.; Rizza, S.A.; O’Brien, D.; et al. Prime, Shock, and Kill: Priming CD4 T Cells from HIV Patients with a BCL-2 Antagonist before HIV Reactivation Reduces HIV Reservoir Size. J. Virol. 2016, 90, 4032–4048. [Google Scholar] [CrossRef]
- Gavegnano, C.; Brehm, J.H.; Dupuy, F.P.; Talla, A.; Ribeiro, S.P.; Kulpa, D.A.; Cameron, C.; Santos, S.; Hurwitz, S.J.; Marconi, V.C.; et al. Novel mechanisms to inhibit HIV reservoir seeding using Jak inhibitors. PLoS Pathog. 2017, 13, e1006740. [Google Scholar] [CrossRef]
- Li, P.; Kaiser, P.; Lampiris, H.W.; Kim, P.; Yukl, S.A.; Havlir, D.V.; Greene, W.C.; Wong, J.K. Stimulating the RIG-I pathway to kill cells in the latent HIV reservoir following viral reactivation. Nat. Med. 2016, 22, 807–811. [Google Scholar] [CrossRef]
- Garcia-Vidal, E.; Castellvi, M.; Pujantell, M.; Badia, R.; Jou, A.; Gomez, L.; Puig, T.; Clotet, B.; Ballana, E.; Riveira-Munoz, E.; et al. Evaluation of the Innate Immune Modulator Acitretin as a Strategy To Clear the HIV Reservoir. Antimicrob. Agents Chemother. 2017, 61, e01368-17. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Bruckman, R.S.; Chu, Y.L.; Trout, R.N.; Spector, S.A. SMAC Mimetics Induce Autophagy-Dependent Apoptosis of HIV-1-Infected Resting Memory CD4+ T Cells. Cell Host Microbe 2018, 24, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Hussein, M.; Molina, M.A.; Berkhout, B.; Herrera-Carrillo, E. A CRISPR-Cas Cure for HIV/AIDS. Int. J. Mol. Sci. 2023, 24, 1563. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Approach | Class | Mechanism | Examples | Section | |
|---|---|---|---|---|---|
| Latency-Promoting Agents | RNA-induced silencing | si/shRNA | RNA-induced silencing | PromA; LTR-362; S4-siRNA | Section 3.1 |
| lncRNA | NEAT1; NRON; PVT1; NKILA; AK130181 | Section 3.1 | |||
| Tat inhibition | trans-dominant Tat mutant | Inhibition of Tat function | Nullbasic | Section 3.2 | |
| Tat inhibitor | Didehydro-cortistatin A (dCA) | Section 3.2 | |||
| Latency-Reversing Agents | Epigenetic modifiers | lncRNA | RNA-induced gene expression | HEAL; MALAT1 | Section 4.1.1 |
| Histone deacetylase inhibitors (HDACi) | Inhibition of histone deacetylases | Valproic acid; Vorinostat (SAHA); Panobinostat; Romidepsin; Givinosat; Belinostat; Entinostat; CC-4a | Section 4.1.2 | ||
| Histone methyltransferases inhibitors (HMTi) | Modification of histone methylation | Chaetocin; DZNep; BIX-01294 | Section 4.1.3 | ||
| Bromodomain and Extra-Terminal Domain Inhibitors (BETi) | P-TEFb release | JQ1; I-BET151; OTX015; CPI-203; MMQO; RVX-208 | Section 4.1.4 | ||
| PAF1C inhibitor | Release of promoter-proximal paused RNA Pol II | iPAF1C | Section 4.3 | ||
| Activators/inhibitors of Inducible Host Factors | TLR agonists | Activation of the NF-κB, NFAT, and AP-1 pathways | Poly-ICLC; MGN1703; GS-986; GS-9620 (Vesatolimod) | Section 4.2.1 | |
| PKC agonists | Canonical NF-κB activation | PMA; Prostratin; Bryostratin; Ingenol | Section 4.2.2 | ||
| PTEN inhibitor | Disulfiram | Section 4.2.2 | |||
| Smac mimetic/ IAP antagonist | Non-canonical NF-κB activation | SBI-0637142; AZD5582; Ciapavir; Debio1143 (Xevinapant) | Section 4.2.3 | ||
| IL-15 stimulation | JAK/STAT activation | IL-15; N-803 | Section 4.3 | ||
| Benzotriazoles | STAT5 activation | HODHBt | Section 4.3 | ||
| TGF-β inhibitors | TGF-β inhibition | Galunisertib | Section 4.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duggan, N.N.; Dragic, T.; Chanda, S.K.; Pache, L. Breaking the Silence: Regulation of HIV Transcription and Latency on the Road to a Cure. Viruses 2023, 15, 2435. https://doi.org/10.3390/v15122435
Duggan NN, Dragic T, Chanda SK, Pache L. Breaking the Silence: Regulation of HIV Transcription and Latency on the Road to a Cure. Viruses. 2023; 15(12):2435. https://doi.org/10.3390/v15122435
Chicago/Turabian StyleDuggan, Natasha N., Tatjana Dragic, Sumit K. Chanda, and Lars Pache. 2023. "Breaking the Silence: Regulation of HIV Transcription and Latency on the Road to a Cure" Viruses 15, no. 12: 2435. https://doi.org/10.3390/v15122435
APA StyleDuggan, N. N., Dragic, T., Chanda, S. K., & Pache, L. (2023). Breaking the Silence: Regulation of HIV Transcription and Latency on the Road to a Cure. Viruses, 15(12), 2435. https://doi.org/10.3390/v15122435