
An Inducible ESCRT-III Inhibition Tool to Control HIV-1 Budding

 , , ,
, , ,  and
and 
Abstract
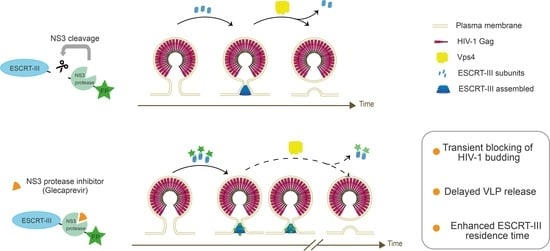
{kind=link}
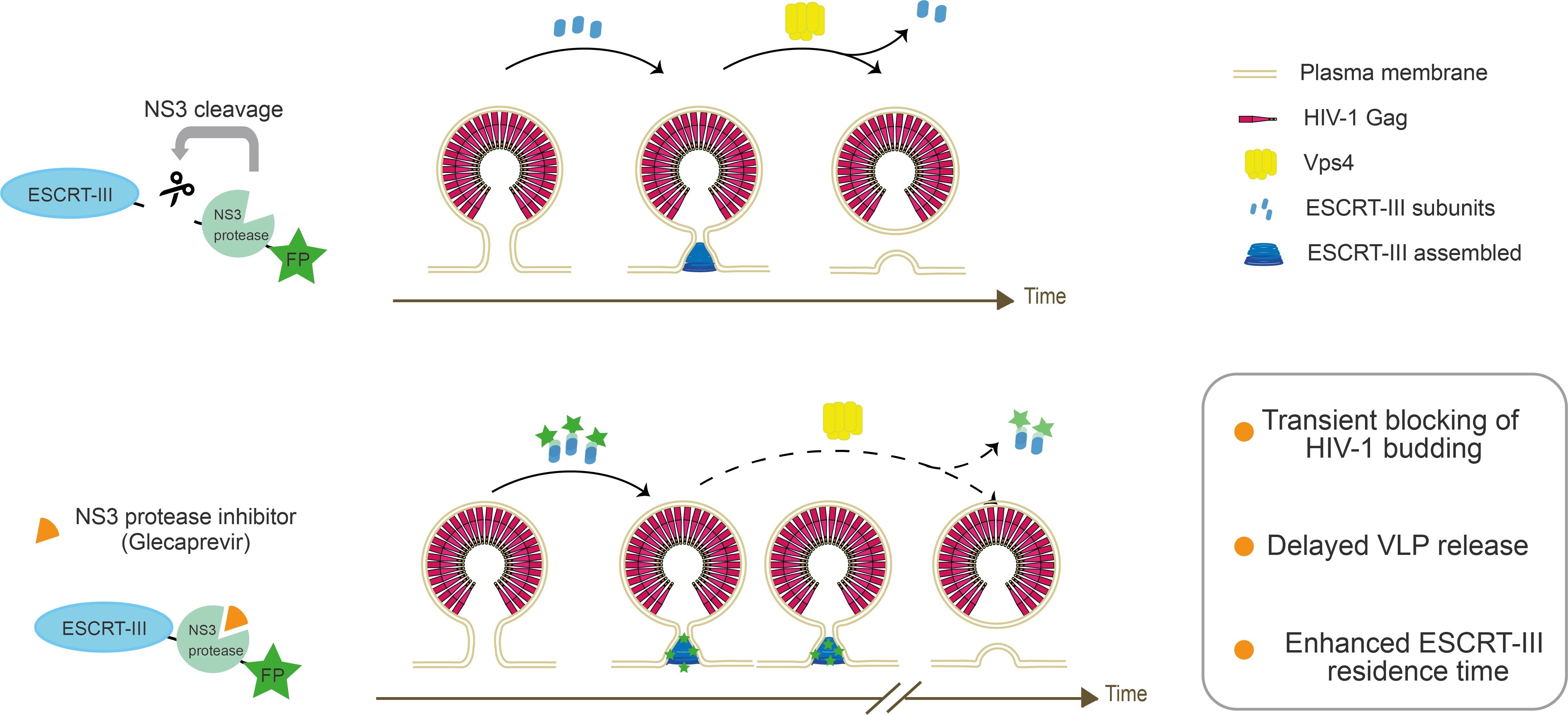
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. DNA Constructs
2.2. Cells Culture, Transfection and Immunoblotting Assay
2.3. Image Acquisition and Analysis
2.4. Single Particle Tracking
3. Results
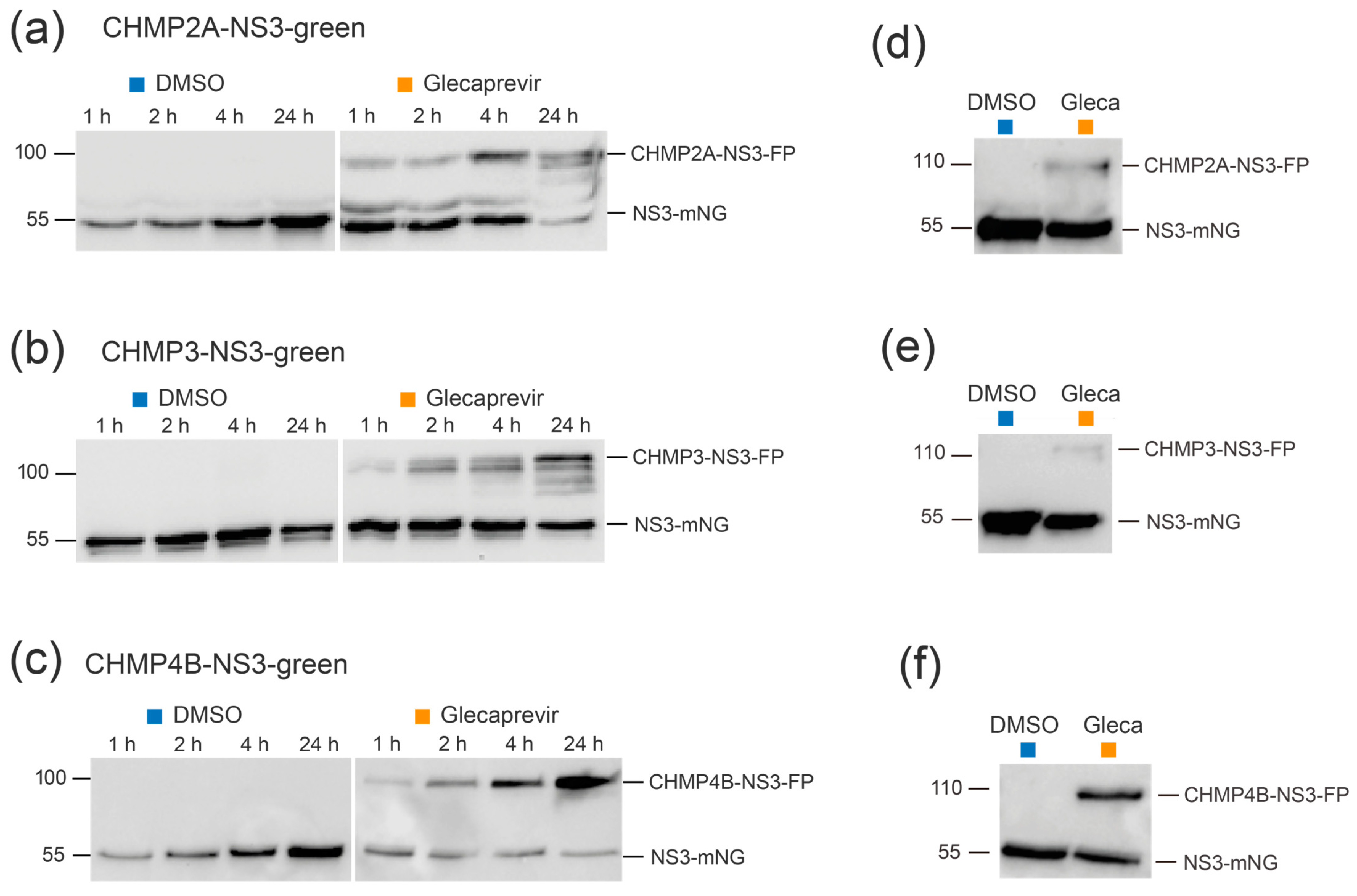
3.1. Expression and Autocleavage of CHMP-NS3-FP
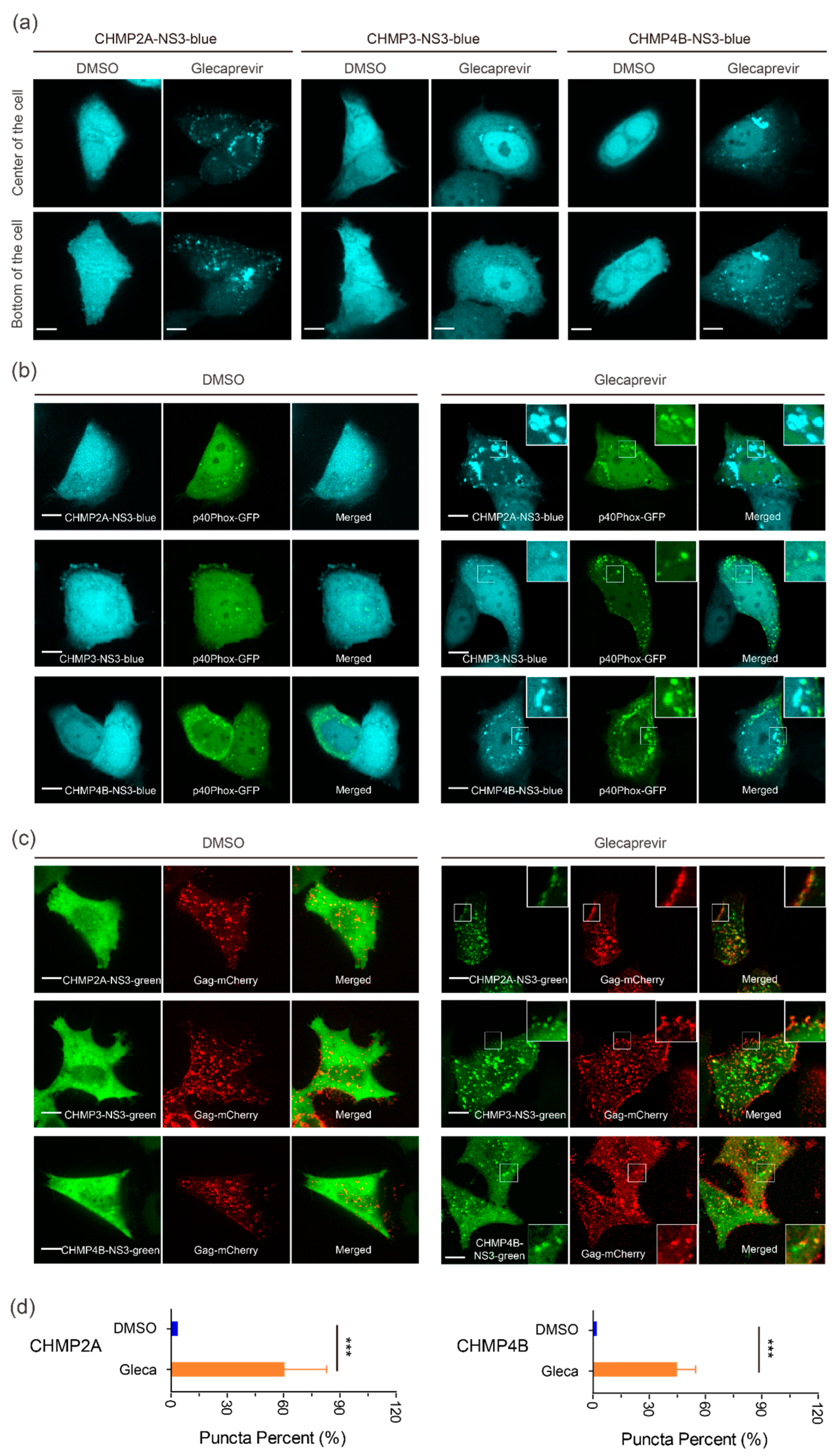
3.2. Cellular Localization of CHMP-NS3-FP Proteins
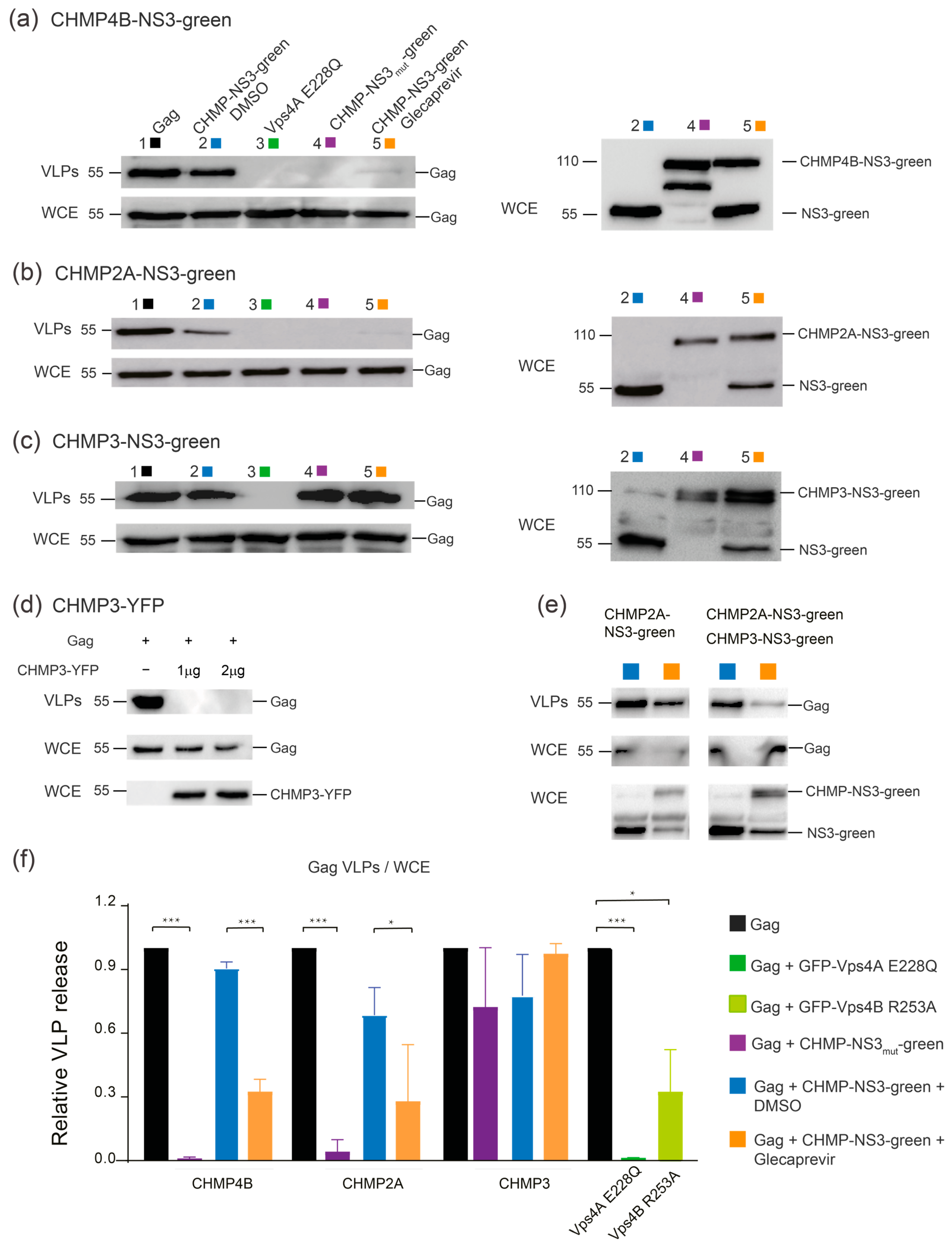
3.3. Partial Inhibition of Gag VLP Release by CHMP-NS3-FP Proteins
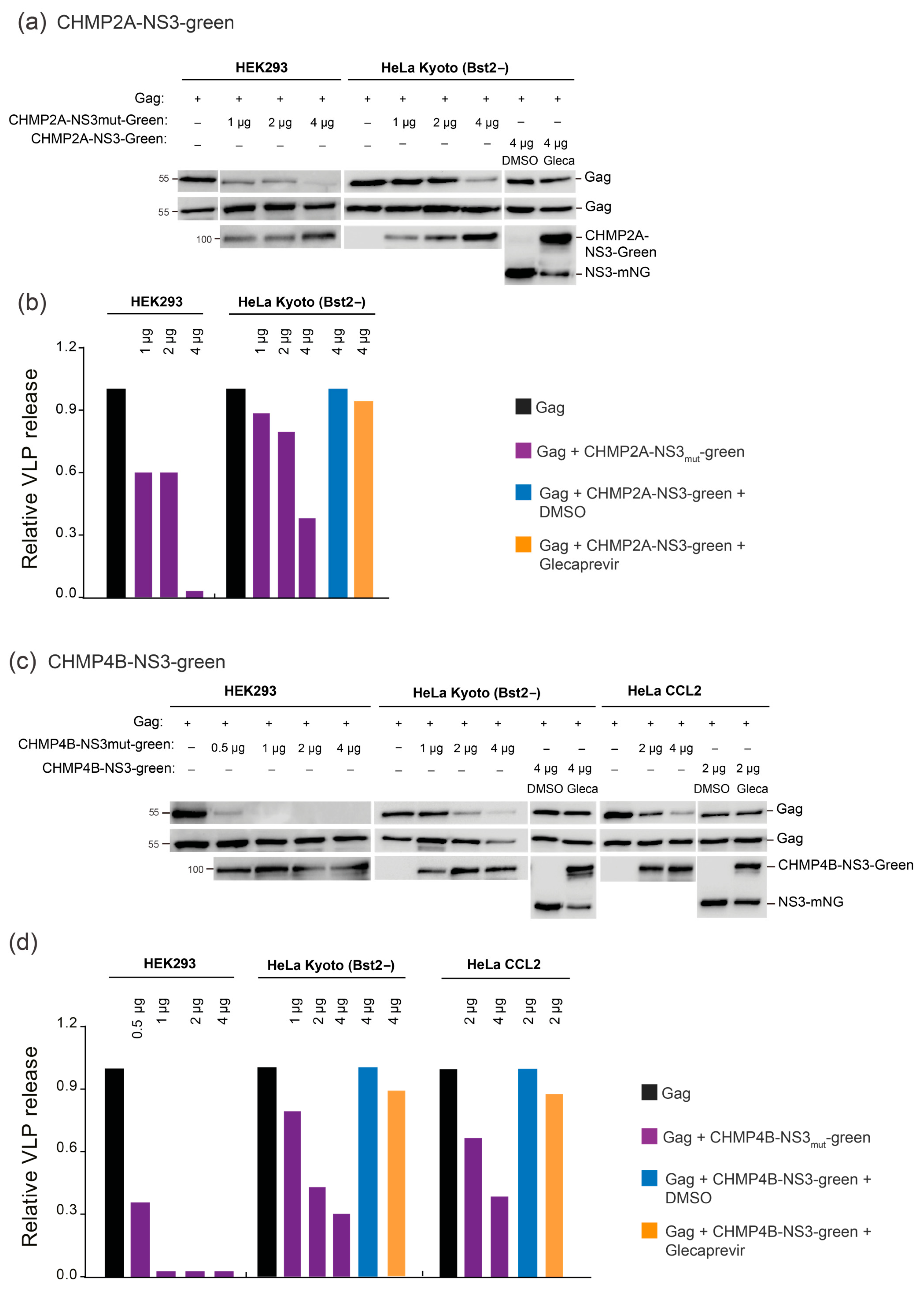
3.4. Cell-Line-Specific and Dose-Dependent Effects of CHMP-NS3-FP Proteins
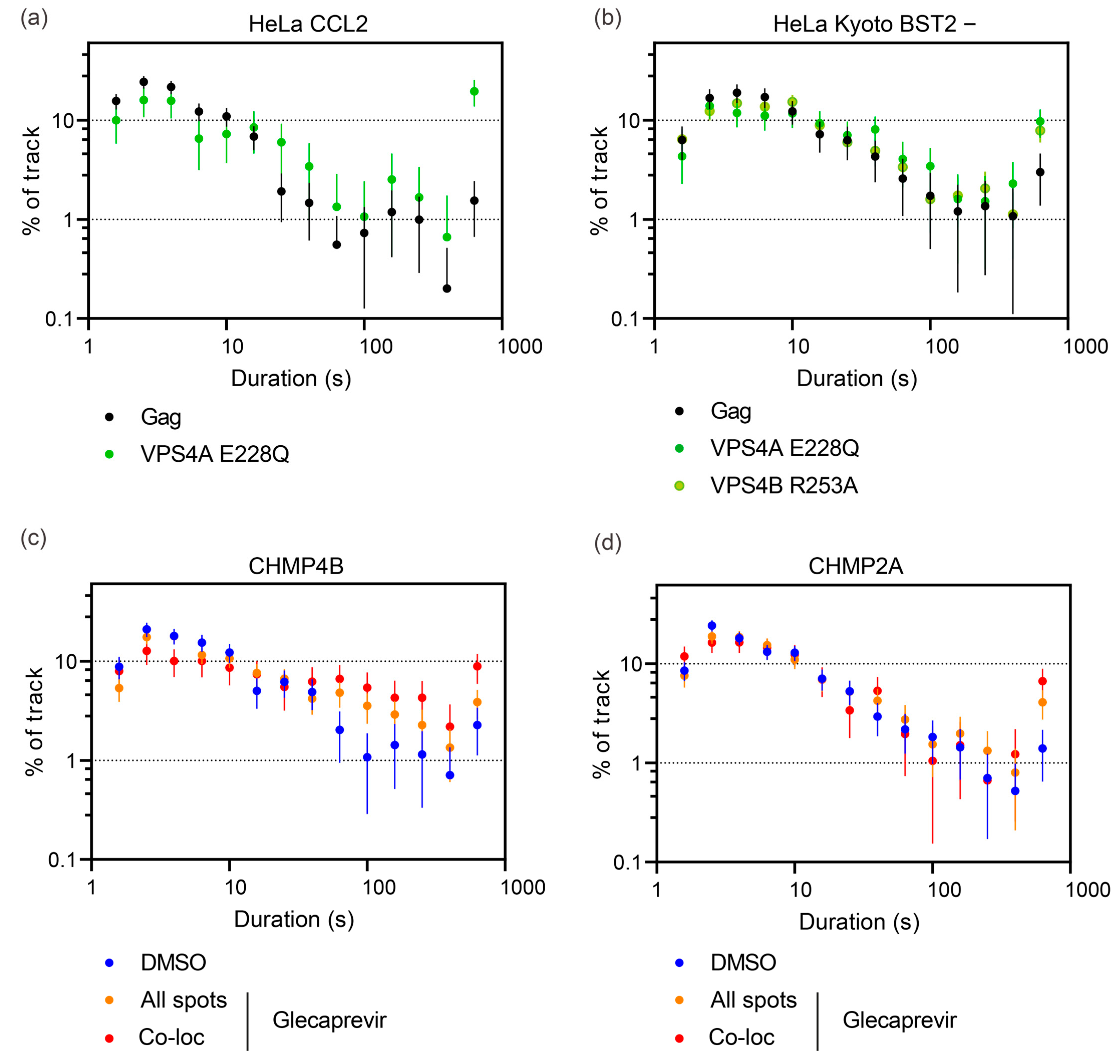
3.5. Quantitative Analysis of HIV-1 Gag VLP Dynamics Reveals Drug-Dependent Prolongation of Gag VLP Retention
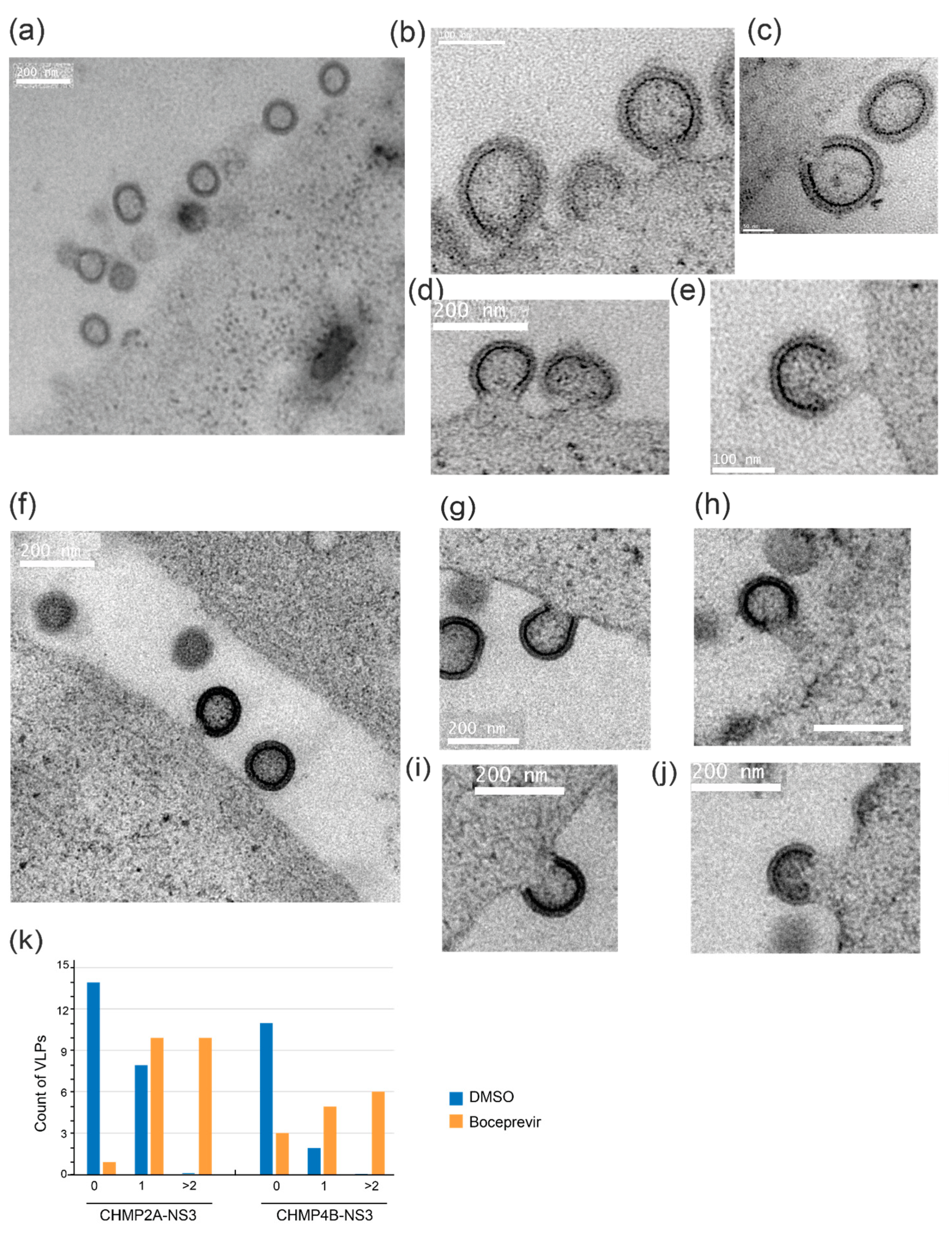
3.6. Electron Microscopy Reveals Drug-Induced Impairment of Late-Stage HIV-1 VLP Budding
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jouvenet, N.; Neil, S.J.; Bess, C.; Johnson, M.C.; Virgen, C.A.; Simon, S.M.; Bieniasz, P.D. Plasma membrane is the site of productive HIV-1 particle assembly. PLoS Biol. 2006, 4, e435. [Google Scholar] [CrossRef] [PubMed]
- Finzi, A.; Orthwein, A.; Mercier, J.; Cohen, E.A. Productive human immunodeficiency virus type 1 assembly takes place at the plasma membrane. J. Virol. 2007, 81, 7476–7490. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ganser-Pornillos, B.K.; Yeager, M.; Sundquist, W.I. The structural biology of HIV assembly. Curr. Opin. Struct. Biol. 2008, 18, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Morita, E.; Sundquist, W.I. Retrovirus budding. Annu. Rev. Cell Dev. Biol. 2004, 20, 395–425. [Google Scholar] [CrossRef] [PubMed]
- Carlton, J.G.; Martin-Serrano, J. Parallels between cytokinesis and retroviral budding: A role for the ESCRT machinery. Science 2007, 316, 1908–1912. [Google Scholar] [CrossRef] [PubMed]
- Welsch, S.; Muller, B.; Krausslich, H.G. More than one door—Budding of enveloped viruses through cellular membranes. FEBS Lett. 2007, 581, 2089–2097. [Google Scholar] [CrossRef]
- Fujii, K.; Hurley, J.H.; Freed, E.O. Beyond Tsg101: The role of Alix in ‘ESCRTing’ HIV-1. Nat. Rev. Microbiol. 2007, 5, 912–916. [Google Scholar] [CrossRef]
- Bieniasz, P.D. The cell biology of HIV-1 virion genesis. Cell Host Microbe 2009, 5, 550–558. [Google Scholar] [CrossRef]
- Gheysen, D.; Jacobs, E.; de Foresta, F.; Thiriart, C.; Francotte, M.; Thines, D.; De Wilde, M. Assembly and release of HIV-1 precursor Pr55gag virus-like particles from recombinant baculovirus-infected insect cells. Cell 1989, 59, 103–112. [Google Scholar] [CrossRef]
- Gottlinger, H.; Dorfman, T.; Sodroski, J.; Haseltine, W. Effect of mutations affecting the p6 gag protein on human immunodeficiency virus particle release. Proc. Natl. Acad. Sci. USA 1991, 88, 3195–3199. [Google Scholar] [CrossRef]
- Parent, L.; Bennett, R.; Craven, R.; Nelle, T.; Krishna, N.; Bowzard, J.; Wilson, C.; Puffer, B.; Montelaro, R.; Wills, J. Positionally independent and exchangeable late budding functions of the Rous sarcoma virus and human immunodeficiency virus Gag proteins. J. Virol. 1995, 69, 5455–5460. [Google Scholar] [CrossRef] [PubMed]
- Strack, B.; Calistri, A.; Gottlinger, H.G. Late assembly domain function can exhibit context dependence and involves ubiquitin residues implicated in endocytosis. J. Virol. 2002, 76, 5472–5479. [Google Scholar] [CrossRef] [PubMed]
- Martin-Serrano, J.; Perez-Caballero, D.; Bieniasz, P.D. Context-dependent effects of L domains and ubiquitination on viral budding. J. Virol. 2004, 78, 5554–5563. [Google Scholar] [CrossRef]
- Garrus, J.; von Schwedler, U.; Pornillos, O.; Morham, S.; Zavitz, K.; Wang, H.; Wettstein, D.; Stray, K.; Cote, M.; Rich, R. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 2001, 107, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Martin-Serrano, J.; Zang, T.; Bieniasz, P. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat. Med. 2001, 7, 1313–1319. [Google Scholar] [CrossRef]
- VerPlank, L.; Bouamr, F.; LaGrassa, T.; Agresta, B.; Kikonyogo, A.; Leis, J.; Carter, C. Tsg101, a homologue of ubiquitin-conjugating (E2) enzymes, binds the L domain in HIV type 1 Pr55(Gag). Proc. Natl. Acad. Sci. USA 2001, 98, 7724–7729. [Google Scholar] [CrossRef]
- Strack, B.; Calistri, A.; Popova, E.; Gottlinger, H. AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell 2003, 114, 689–699. [Google Scholar] [CrossRef]
- von Schwedler, U.K.; Stuchell, M.; Muller, B.; Ward, D.M.; Chung, H.Y.; Morita, E.; Wang, H.E.; Davis, T.; He, G.P.; Cimbora, D.M.; et al. The protein network of HIV budding. Cell 2003, 114, 701–713. [Google Scholar] [CrossRef]
- Martin-Serrano, J.; Yarovoy, A.; Perez-Caballero, D.; Bieniasz, P.D. Divergent retroviral late-budding domains recruit vacuolar protein sorting factors by using alternative adaptor proteins. Proc. Natl. Acad. Sci. USA 2003, 100, 12414–12419. [Google Scholar] [CrossRef]
- Vincent, O.; Rainbow, L.; Tilburn, J.; Arst, H.N., Jr.; Penalva, M.A. YPXL/I is a protein interaction motif recognized by aspergillus PalA and its human homologue, AIP1/Alix. Mol. Cell Biol. 2003, 23, 1647–1655. [Google Scholar] [CrossRef]
- Henne, W.M.; Stenmark, H.; Emr, S.D. Molecular mechanisms of the membrane sculpting ESCRT pathway. Cold Spring Harb. Perspect. Biol. 2013, 5, a016766. [Google Scholar] [CrossRef] [PubMed]
- Scourfield, E.J.; Martin-Serrano, J. Growing functions of the ESCRT machinery in cell biology and viral replication. Biochem. Soc. Trans. 2017, 45, 613–634. [Google Scholar] [CrossRef] [PubMed]
- McCullough, J.; Frost, A.; Sundquist, W.I. Structures, Functions, and Dynamics of ESCRT-III/Vps4 Membrane Remodeling and Fission Complexes. Annu. Rev. Cell Dev. Biol. 2018, 34, 85–109. [Google Scholar] [CrossRef] [PubMed]
- Schoneberg, J.; Lee, I.H.; Iwasa, J.H.; Hurley, J.H. Reverse-topology membrane scission by the ESCRT proteins. Nat. Rev. Mol. Cell Biol. 2017, 18, 5–17. [Google Scholar] [CrossRef]
- Caillat, C.; Maity, S.; Miguet, N.; Roos, W.H.; Weissenhorn, W. The role of VPS4 in ESCRT-III polymer remodeling. Biochem. Soc. Trans. 2019, 47, 441–448. [Google Scholar] [CrossRef]
- Muziol, T.; Pineda-Molina, E.; Ravelli, R.B.; Zamborlini, A.; Usami, Y.; Gottlinger, H.; Weissenhorn, W. Structural basis for budding by the ESCRT-III factor CHMP3. Dev. Cell 2006, 10, 821–830. [Google Scholar] [CrossRef]
- Bajorek, M.; Schubert, H.L.; McCullough, J.; Langelier, C.; Eckert, D.M.; Stubblefield, W.M.; Uter, N.T.; Myszka, D.G.; Hill, C.P.; Sundquist, W.I. Structural basis for ESCRT-III protein autoinhibition. Nat. Struct. Mol. Biol. 2009, 16, 754–762. [Google Scholar] [CrossRef]
- Zamborlini, A.; Usami, Y.; Radoshitzky, S.R.; Popova, E.; Palu, G.; Gottlinger, H. Release of autoinhibition converts ESCRT-III components into potent inhibitors of HIV-1 budding. Proc. Natl. Acad. Sci. USA 2006, 103, 19140–19145. [Google Scholar] [CrossRef]
- Shim, S.; Kimpler, L.A.; Hanson, P.I. Structure/Function Analysis of Four Core ESCRT-III Proteins Reveals Common Regulatory Role for Extreme C-Terminal Domain. Traffic 2007, 8, 1068–1079. [Google Scholar] [CrossRef]
- Lata, S.; Roessle, M.; Solomons, J.; Jamin, M.; Gottlinger, H.G.; Svergun, D.I.; Weissenhorn, W. Structural basis for autoinhibition of ESCRT-III CHMP3. J. Mol. Biol. 2008, 378, 818–827. [Google Scholar] [CrossRef]
- McCullough, J.; Clippinger, A.K.; Talledge, N.; Skowyra, M.L.; Saunders, M.G.; Naismith, T.V.; Colf, L.A.; Afonine, P.; Arthur, C.; Sundquist, W.I.; et al. Structure and membrane remodeling activity of ESCRT-III helical polymers. Science 2015, 350, 1548–1551. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Henne, W.M.; Borbat, P.P.; Buchkovich, N.J.; Freed, J.H.; Mao, Y.; Fromme, J.C.; Emr, S.D. Structural basis for activation, assembly and membrane binding of ESCRT-III Snf7 filaments. Elife 2015, 4, e12548. [Google Scholar] [CrossRef] [PubMed]
- McMillan, B.J.; Tibbe, C.; Jeon, H.; Drabek, A.A.; Klein, T.; Blacklow, S.C. Electrostatic Interactions between Elongated Monomers Drive Filamentation of Drosophila Shrub, a Metazoan ESCRT-III Protein. Cell Rep. 2016, 16, 1211–1217. [Google Scholar] [CrossRef]
- Azad, K.; Guilligay, D.; Boscheron, C.; Maity, S.; De Franceschi, N.; Sulbaran, G.; Effantin, G.; Wang, H.; Kleman, J.P.; Bassereau, P.; et al. Structural basis of CHMP2A–CHMP3 ESCRT-III polymer assembly and membrane cleavage. Nat. Struct. Mol. Biol. 2023, 30, 81–90. [Google Scholar] [CrossRef]
- Lata, S.; Schoehn, G.; Jain, A.; Pires, R.; Piehler, J.; Gottlinger, H.G.; Weissenhorn, W. Helical structures of ESCRT-III are disassembled by VPS4. Science 2008, 321, 1354–1357. [Google Scholar] [CrossRef] [PubMed]
- Pires, R.; Hartlieb, B.; Signor, L.; Schoehn, G.; Lata, S.; Roessle, M.; Moriscot, C.; Popov, S.; Hinz, A.; Jamin, M.; et al. A crescent-shaped ALIX dimer targets ESCRT-III CHMP4 filaments. Structure 2009, 17, 843–856. [Google Scholar] [CrossRef]
- Mierzwa, B.E.; Chiaruttini, N.; Redondo-Morata, L.; von Filseck, J.M.; Konig, J.; Larios, J.; Poser, I.; Muller-Reichert, T.; Scheuring, S.; Roux, A.; et al. Dynamic subunit turnover in ESCRT-III assemblies is regulated by Vps4 to mediate membrane remodelling during cytokinesis. Nat. Cell Biol. 2017, 19, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Bertin, A.; de Franceschi, N.; de la Mora, E.; Maity, S.; Alqabandi, M.; Miguet, N.; di Cicco, A.; Roos, W.H.; Mangenot, S.; Weissenhorn, W.; et al. Human ESCRT-III polymers assemble on positively curved membranes and induce helical membrane tube formation. Nat. Commun. 2020, 11, 2663. [Google Scholar] [CrossRef]
- Moser von Filseck, J.; Barberi, L.; Talledge, N.; Johnson, I.E.; Frost, A.; Lenz, M.; Roux, A. Anisotropic ESCRT-III architecture governs helical membrane tube formation. Nat. Commun. 2020, 11, 1516. [Google Scholar] [CrossRef] [PubMed]
- Henne, W.M.; Buchkovich, N.J.; Zhao, Y.; Emr, S.D. The Endosomal Sorting Complex ESCRT-II Mediates the Assembly and Architecture of ESCRT-III Helices. Cell 2012, 151, 356–371. [Google Scholar] [CrossRef]
- Hanson, P.I.; Roth, R.; Lin, Y.; Heuser, J.E. Plasma membrane deformation by circular arrays of ESCRT-III protein filaments. J. Cell Biol. 2008, 180, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Cashikar, A.G.; Shim, S.; Roth, R.; Maldazys, M.R.; Heuser, J.E.; Hanson, P.I. Structure of cellular ESCRT-III spirals and their relationship to HIV budding. Elife 2014, 3, e02184. [Google Scholar] [CrossRef] [PubMed]
- Bodon, G.; Chassefeyre, R.; Pernet-Gallay, K.; Martinelli, N.; Effantin, G.; Lutje Hulsik, D.; Belly, A.; Goldberg, Y.; Chatellard-Causse, C.; Blot, B.; et al. Charged Multivesicular Body Protein 2B (CHMP2B) of the Endosomal Sorting Complex Required for Transport-III (ESCRT-III) Polymerizes into Helical Structures Deforming the Plasma Membrane. J. Biol. Chem. 2011, 286, 40276–40286. [Google Scholar] [CrossRef]
- Guizetti, J.; Schermelleh, L.; Mantler, J.; Maar, S.; Poser, I.; Leonhardt, H.; Muller-Reichert, T.; Gerlich, D.W. Cortical Constriction During Abscission Involves Helices of ESCRT-III-Dependent Filaments. Science 2011, 331, 1616–1620. [Google Scholar] [CrossRef] [PubMed]
- Goliand, I.; Adar-Levor, S.; Segal, I.; Nachmias, D.; Dadosh, T.; Kozlov, M.M.; Elia, N. Resolving ESCRT-III Spirals at the Intercellular Bridge of Dividing Cells Using 3D STORM. Cell Rep. 2018, 24, 1756–1764. [Google Scholar] [CrossRef]
- Wenzel, D.M.; Mackay, D.R.; Skalicky, J.J.; Paine, E.L.; Miller, M.S.; Ullman, K.S.; Sundquist, W.I. Comprehensive analysis of the human ESCRT-III-MIT domain interactome reveals new cofactors for cytokinetic abscission. Elife 2022, 11, e77779. [Google Scholar] [CrossRef]
- Adell, M.A.Y.; Migliano, S.M.; Upadhyayula, S.; Bykov, Y.S.; Sprenger, S.; Pakdel, M.; Vogel, G.F.; Jih, G.; Skillern, W.; Behrouzi, R.; et al. Recruitment dynamics of ESCRT-III and Vps4 to endosomes and implications for reverse membrane budding. Elife 2017, 6, e31652. [Google Scholar] [CrossRef]
- Maity, S.; Caillat, C.; Miguet, N.; Sulbaran, G.; Effantin, G.; Schoehn, G.; Roos, W.H.; Weissenhorn, W. VPS4 triggers constriction and cleavage of ESCRT-III helical filaments. Sci. Adv. 2019, 5, eaau7198. [Google Scholar] [CrossRef]
- Kieffer, C.; Skalicky, J.J.; Morita, E.; De Domenico, I.; Ward, D.M.; Kaplan, J.; Sundquist, W.I. Two distinct modes of ESCRT-III recognition are required for VPS4 functions in lysosomal protein targeting and HIV-1 budding. Dev. Cell 2008, 15, 62–73. [Google Scholar] [CrossRef]
- Fabrikant, G.; Lata, S.; Riches, J.D.; Briggs, J.A.; Weissenhorn, W.; Kozlov, M.M. Computational model of membrane fission catalyzed by ESCRT-III. PLoS Comput. Biol. 2009, 5, e1000575. [Google Scholar] [CrossRef]
- Rheinemann, L.; Downhour, D.M.; Bredbenner, K.; Mercenne, G.; Davenport, K.A.; Schmitt, P.T.; Necessary, C.R.; McCullough, J.; Schmitt, A.P.; Simon, S.M.; et al. RetroCHMP3 blocks budding of enveloped viruses without blocking cytokinesis. Cell 2021, 184, 5419–5431.e16. [Google Scholar] [CrossRef] [PubMed]
- Morita, E.; Sandrin, V.; McCullough, J.; Katsuyama, A.; Baci Hamilton, I.; Sundquist, W.I. ESCRT-III Protein Requirements for HIV-1 Budding. Cell Host Microbe 2011, 9, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Effantin, G.; Dordor, A.; Sandrin, V.; Martinelli, N.; Sundquist, W.I.; Schoehn, G.; Weissenhorn, W. ESCRT-III CHMP2A and CHMP3 form variable helical polymers in vitro and act synergistically during HIV-1 budding. Cell. Microbiol. 2013, 15, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Jouvenet, N.; Bieniasz, P.D.; Simon, S.M. Imaging the biogenesis of individual HIV-1 virions in live cells. Nature 2008, 454, 236–240. [Google Scholar] [CrossRef]
- Jouvenet, N.; Zhadina, M.; Bieniasz, P.D.; Simon, S.M. Dynamics of ESCRT protein recruitment during retroviral assembly. Nat. Cell Biol. 2011, 13, 394–401. [Google Scholar] [CrossRef]
- Baumgartel, V.; Ivanchenko, S.; Dupont, A.; Sergeev, M.; Wiseman, P.W.; Krausslich, H.G.; Brauchle, C.; Muller, B.; Lamb, D.C. Live-cell visualization of dynamics of HIV budding site interactions with an ESCRT component. Nat. Cell Biol. 2011, 13, 469–474. [Google Scholar] [CrossRef]
- Prescher, J.; Baumgartel, V.; Ivanchenko, S.; Torrano, A.A.; Brauchle, C.; Muller, B.; Lamb, D.C. Super-resolution imaging of ESCRT-proteins at HIV-1 assembly sites. PLoS Pathog. 2015, 11, e1004677. [Google Scholar] [CrossRef]
- Johnson, D.S.; Bleck, M.; Simon, S.M. Timing of ESCRT-III protein recruitment and membrane scission during HIV-1 assembly. Elife 2018, 7, e36221. [Google Scholar] [CrossRef]
- Bleck, M.; Itano, M.S.; Johnson, D.S.; Thomas, V.K.; North, A.J.; Bieniasz, P.D.; Simon, S.M. Temporal and spatial organization of ESCRT protein recruitment during HIV-1 budding. Proc. Natl. Acad. Sci. USA 2014, 111, 12211–12216. [Google Scholar] [CrossRef]
- Shaner, N.C.; Lambert, G.G.; Chammas, A.; Ni, Y.; Cranfill, P.J.; Baird, M.A.; Sell, B.R.; Allen, J.R.; Day, R.N.; Israelsson, M.; et al. A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat. Methods 2013, 10, 407–409. [Google Scholar] [CrossRef]
- Kremers, G.J.; Goedhart, J.; van Munster, E.B.; Gadella, T.W., Jr. Cyan and yellow super fluorescent proteins with improved brightness, protein folding, and FRET Förster radius. Biochemistry 2006, 45, 6570–6580. [Google Scholar] [CrossRef]
- Kotsopoulou, E.; Kim, V.N.; Kingsman, A.J.; Kingsman, S.M.; Mitrophanous, K.A. A Rev-independent human immunodeficiency virus type 1 (HIV-1)-based vector that exploits a codon-optimized HIV-1 gag-pol gene. J. Virol. 2000, 74, 4839–4852. [Google Scholar] [CrossRef] [PubMed]
- Perez-Caballero, D.; Hatziioannou, T.; Martin-Serrano, J.; Bieniasz, P.D. Human immunodeficiency virus type 1 matrix inhibits and confers cooperativity on gag precursor-membrane interactions. J. Virol. 2004, 78, 9560–9563. [Google Scholar] [CrossRef] [PubMed]
- Caillat, C.; Macheboeuf, P.; Wu, Y.; McCarthy, A.A.; Boeri-Erba, E.; Effantin, G.; Gottlinger, H.G.; Weissenhorn, W.; Renesto, P. Asymmetric ring structure of Vps4 required for ESCRT-III disassembly. Nat. Commun. 2015, 6, 8781. [Google Scholar] [CrossRef]
- Kanai, F.; Liu, H.; Field, S.J.; Akbary, H.; Matsuo, T.; Brown, G.E.; Cantley, L.C.; Yaffe, M.B. The PX domains of p47phox and p40phox bind to lipid products of PI(3)K. Nat. Cell Biol. 2001, 3, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Presle, A.; Fremont, S.; Salles, A.; Commere, P.H.; Sassoon, N.; Berlioz-Torrent, C.; Gupta-Rossi, N.; Echard, A. The viral restriction factor tetherin/BST2 tethers cytokinetic midbody remnants to the cell surface. Curr. Biol. 2021, 31, 2203–2213.e5. [Google Scholar] [CrossRef]
- Larson, D.R.; Johnson, M.C.; Webb, W.W.; Vogt, V.M. Visualization of retrovirus budding with correlated light and electron microscopy. Proc. Natl. Acad. Sci. USA 2005, 102, 15453–15458. [Google Scholar] [CrossRef]
- Mohamed, A.M.T.; Chan, H.; Luhur, J.; Bauda, E.; Gallet, B.; Morlot, C.; Cole, L.; Awad, M.; Crawford, S.; Lyras, D.; et al. Chromosome Segregation and Peptidoglycan Remodeling Are Coordinated at a Highly Stabilized Septal Pore to Maintain Bacterial Spore Development. Dev. Cell 2021, 56, 36–51.e5. [Google Scholar] [CrossRef]
- de Chaumont, F.; Dallongeville, S.; Chenouard, N.; Hervé, N.; Pop, S.; Provoost, T.; Meas-Yedid, V.; Pankajakshan, P.; Lecomte, T.; Le Montagner, Y.; et al. Icy: An open bioimage informatics platform for extended reproducible research. Nat. Methods 2012, 9, 690–696. [Google Scholar] [CrossRef]
- Olivo-Marin, J.-C. Extraction of spots in biological images using multiscale products. Pattern Recognit. 2002, 35, 1989–1996. [Google Scholar] [CrossRef]
- Chenouard, N.; Bloch, I.; Olivo-Marin, J.C. Multiple hypothesis tracking for cluttered biological image sequences. IEEE Trans. Pattern Anal. Mach. Intell. 2013, 35, 2736–3750. [Google Scholar] [CrossRef] [PubMed]
- Neil, S.J.; Eastman, S.W.; Jouvenet, N.; Bieniasz, P.D. HIV-1 Vpu promotes release and prevents endocytosis of nascent retrovirus particles from the plasma membrane. PLoS Pathog. 2006, 2, e39. [Google Scholar] [CrossRef] [PubMed]
- Neil, S.J.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Ivanchenko, S.; Godinez, W.J.; Lampe, M.; Krausslich, H.G.; Eils, R.; Rohr, K.; Brauchle, C.; Muller, B.; Lamb, D.C. Dynamics of HIV-1 assembly and release. PLoS Pathog. 2009, 5, e1000652. [Google Scholar] [CrossRef] [PubMed]
- Usami, Y.; Popov, S.; Popova, E.; Inoue, M.; Weissenhorn, W.; Gottlinger, H.G. The ESCRT pathway and HIV-1 budding. Biochem. Soc. Trans. 2009, 37 Pt 1, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Votteler, J.; Sundquist, W.I. Virus budding and the ESCRT pathway. Cell Host Microbe 2013, 14, 232–241. [Google Scholar] [CrossRef]
- Vietri, M.; Radulovic, M.; Stenmark, H. The many functions of ESCRTs. Nat. Rev. Mol. Cell Biol. 2020, 21, 25–42. [Google Scholar] [CrossRef]
- Lin, M.Z.; Glenn, J.S.; Tsien, R.Y. A drug-controllable tag for visualizing newly synthesized proteins in cells and whole animals. Proc. Natl. Acad. Sci. USA 2008, 105, 7744–7749. [Google Scholar] [CrossRef]
- Lin, M.Z.; Tsien, R.Y. TimeSTAMP tagging of newly synthesized proteins. Curr. Protoc. Protein Sci. 2010, 59, 26.5.1–26.5.11. [Google Scholar] [CrossRef]
- Howard, T.L.; Stauffer, D.R.; Degnin, C.R.; Hollenberg, S.M. CHMP1 functions as a member of a newly defined family of vesicle trafficking proteins. J. Cell Sci. 2001, 114 Pt 13, 2395–2404. [Google Scholar] [CrossRef]
- Martin-Serrano, J.; Bieniasz, P.D. A bipartite late-budding domain in human immunodeficiency virus type 1. J. Virol. 2003, 77, 12373–12377. [Google Scholar] [CrossRef] [PubMed]
- Buchkovich, N.J.; Henne, W.M.; Tang, S.; Emr, S.D. Essential N-terminal insertion motif anchors the ESCRT-III filament during MVB vesicle formation. Dev. Cell 2013, 27, 201–214. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Gallet, B.; Moriscot, C.; Pezet, M.; Chatellard, C.; Kleman, J.-P.; Göttlinger, H.; Weissenhorn, W.; Boscheron, C. An Inducible ESCRT-III Inhibition Tool to Control HIV-1 Budding. Viruses 2023, 15, 2289. https://doi.org/10.3390/v15122289
Wang H, Gallet B, Moriscot C, Pezet M, Chatellard C, Kleman J-P, Göttlinger H, Weissenhorn W, Boscheron C. An Inducible ESCRT-III Inhibition Tool to Control HIV-1 Budding. Viruses. 2023; 15(12):2289. https://doi.org/10.3390/v15122289
Chicago/Turabian StyleWang, Haiyan, Benoit Gallet, Christine Moriscot, Mylène Pezet, Christine Chatellard, Jean-Philippe Kleman, Heinrich Göttlinger, Winfried Weissenhorn, and Cécile Boscheron. 2023. "An Inducible ESCRT-III Inhibition Tool to Control HIV-1 Budding" Viruses 15, no. 12: 2289. https://doi.org/10.3390/v15122289
APA StyleWang, H., Gallet, B., Moriscot, C., Pezet, M., Chatellard, C., Kleman, J.-P., Göttlinger, H., Weissenhorn, W., & Boscheron, C. (2023). An Inducible ESCRT-III Inhibition Tool to Control HIV-1 Budding. Viruses, 15(12), 2289. https://doi.org/10.3390/v15122289