

Characterization of a Putative New Member of the Genus Potyvirus from Kudzu (Pueraria montana var. lobata) in Mississippi

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Double-Stranded RNA Analysis
2.3. Electron Microscopy
2.4. Genome Sequencing, Sequence Assembly, and Analyses
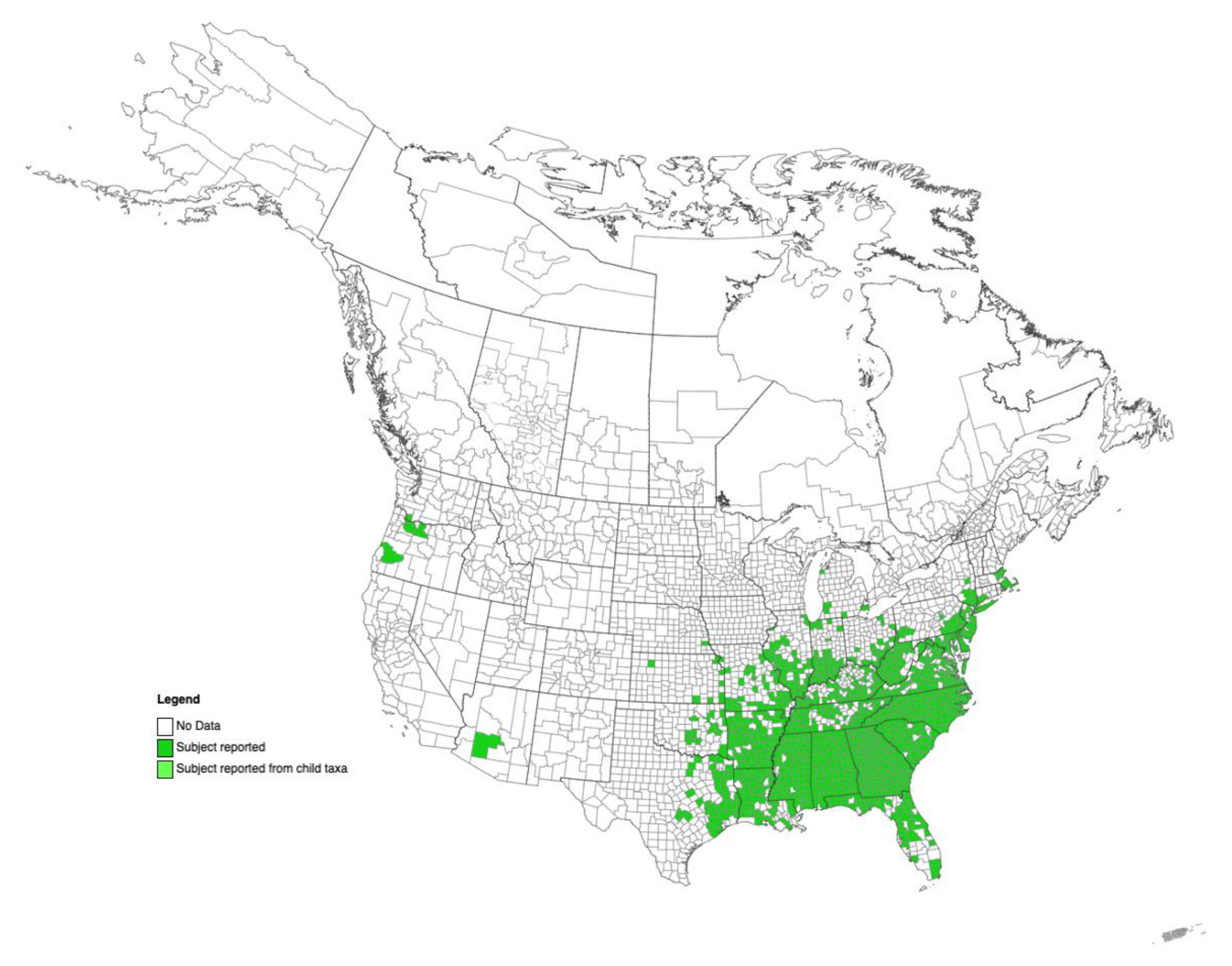
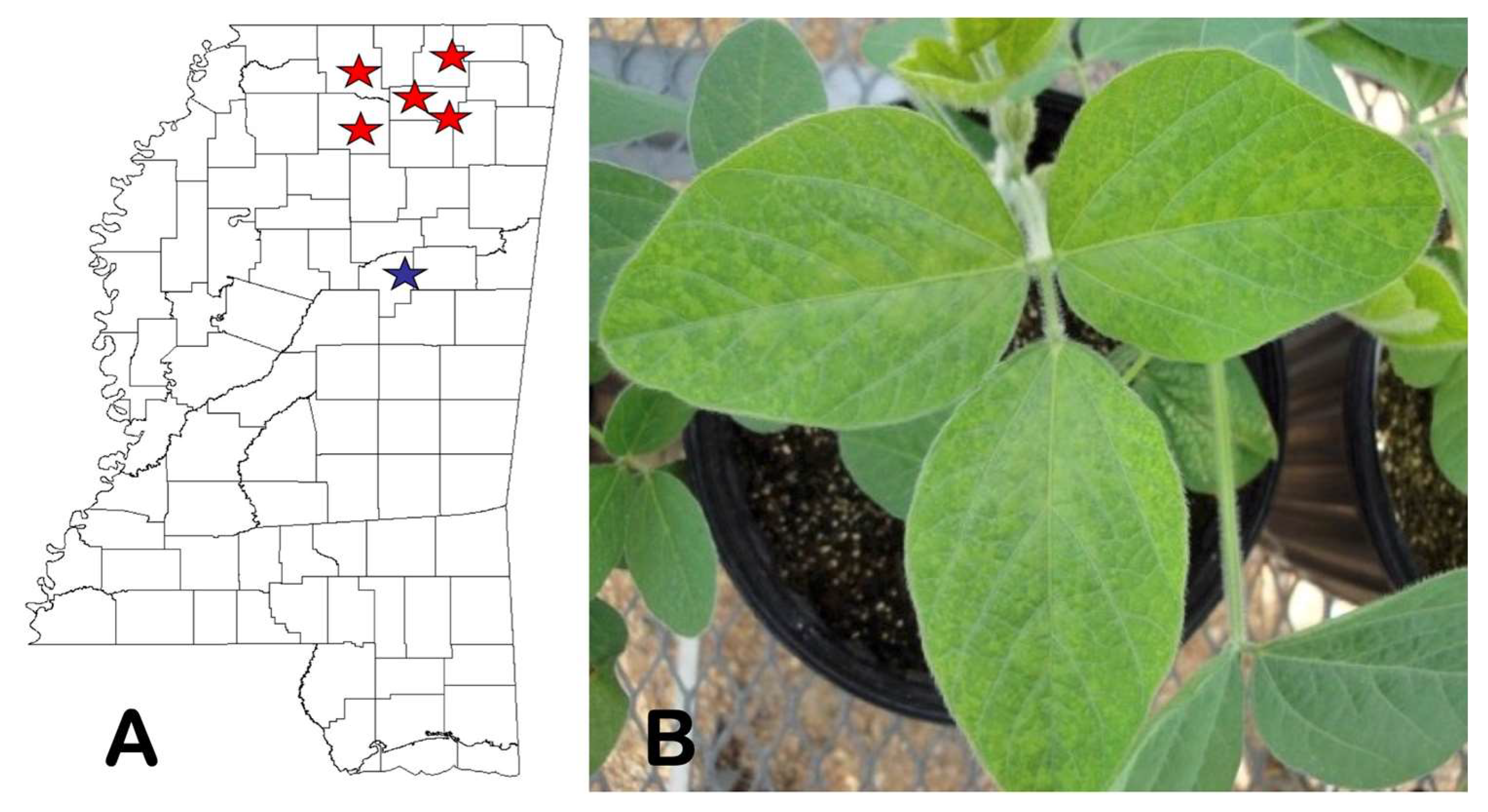
2.5. State-Wide Survey of Kudzu Patches and Search for Alternative Hosts
2.6. Identification of Possible Vector and Transmission Assays
3. Results
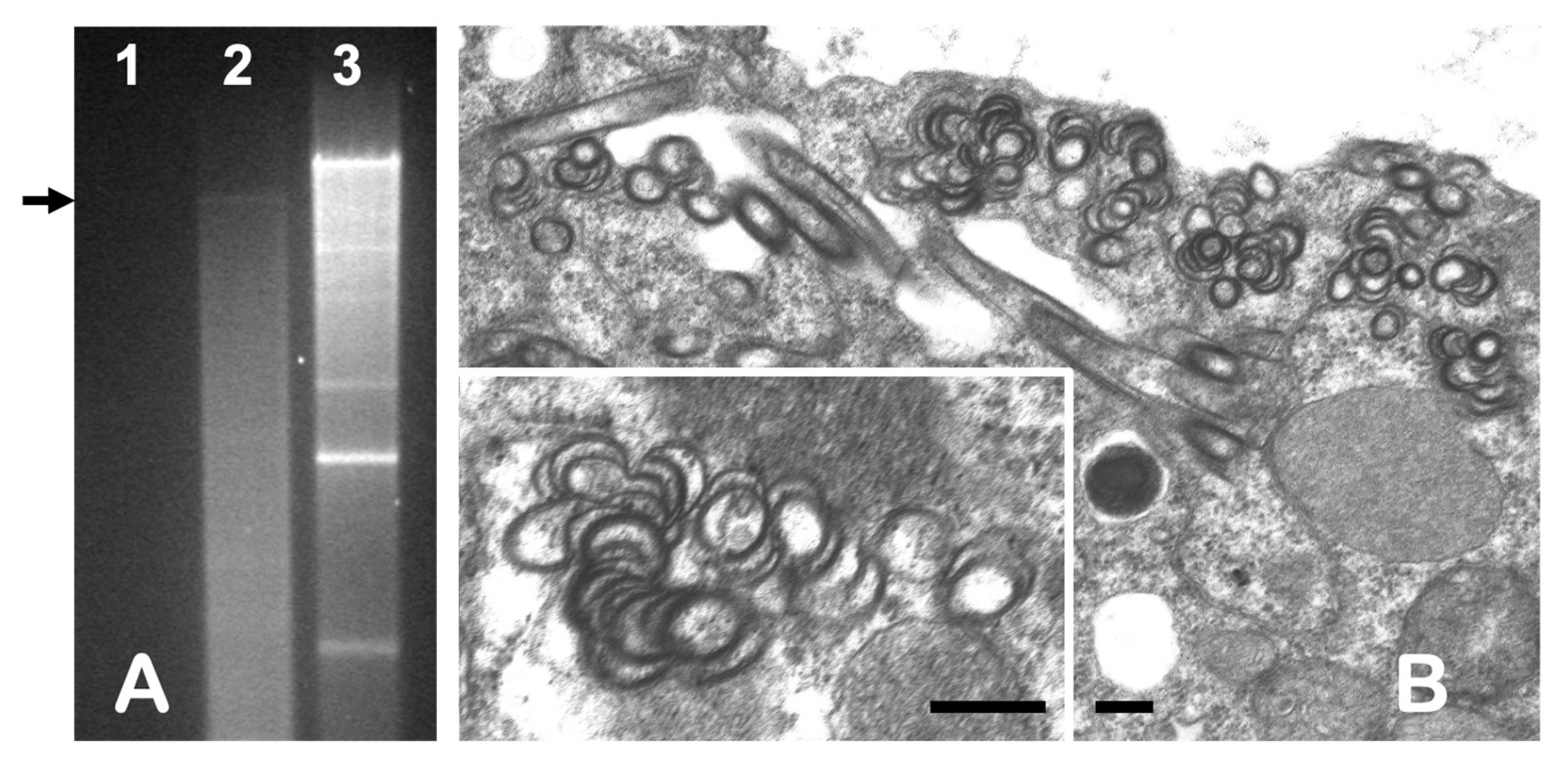
3.1. dsRNA Analysis and Electron Microscopy
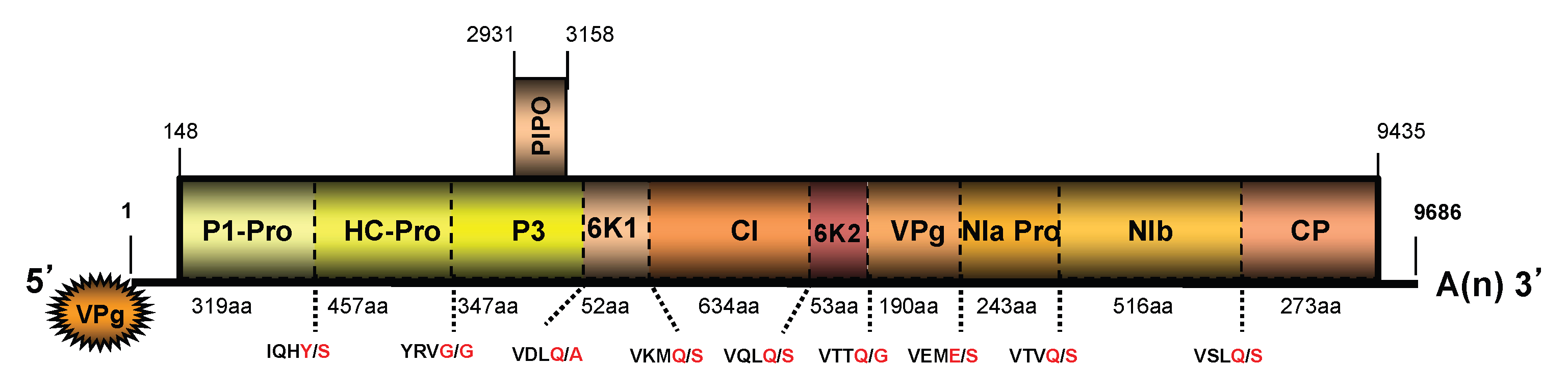
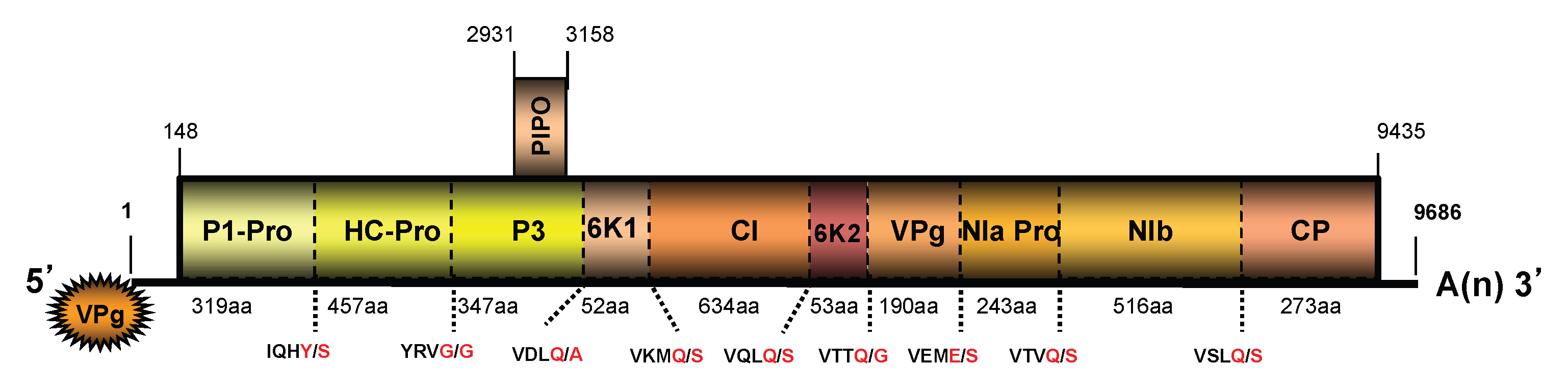
3.2. Genome Sequences of a Virus from Kudzu and Their Analyses
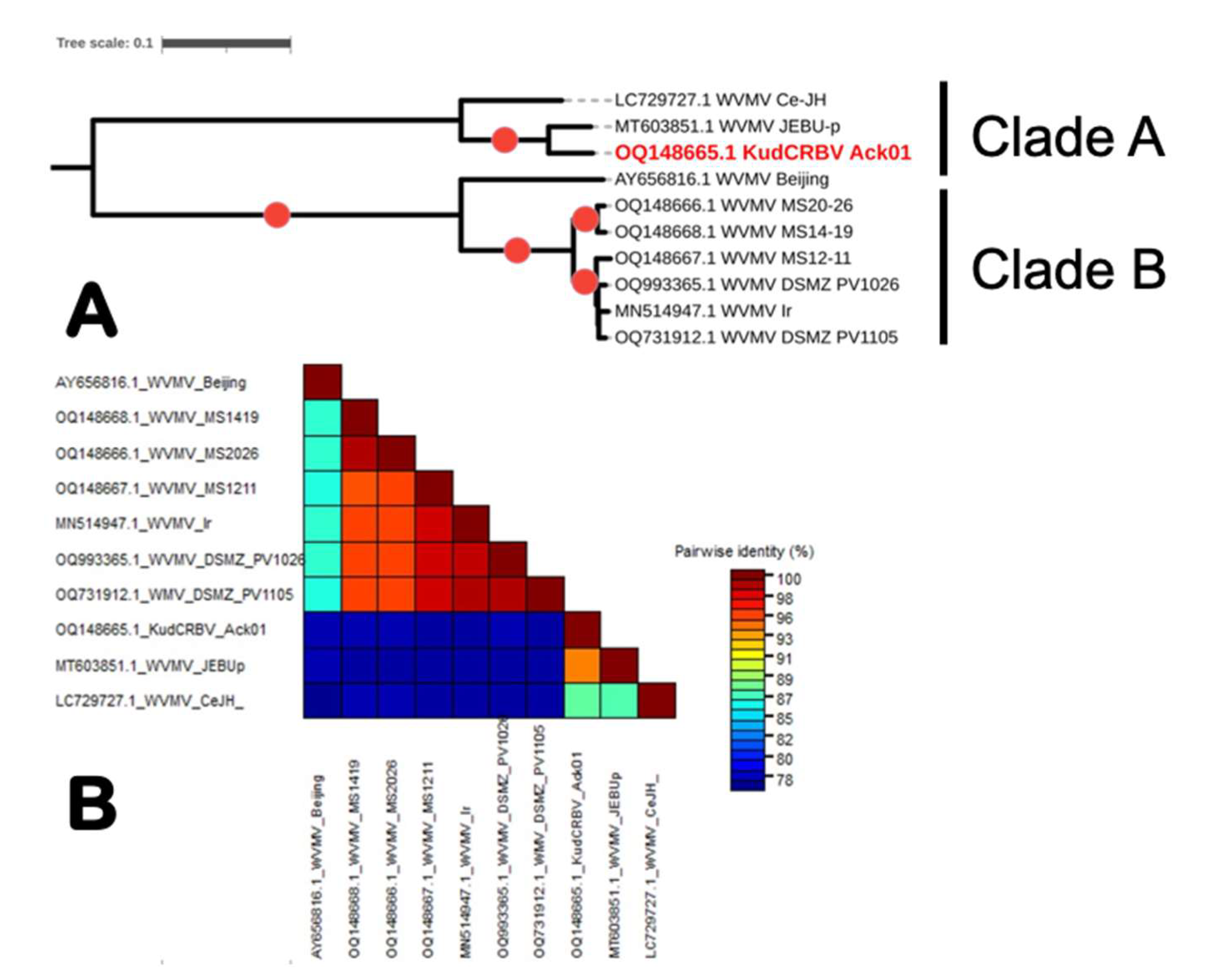
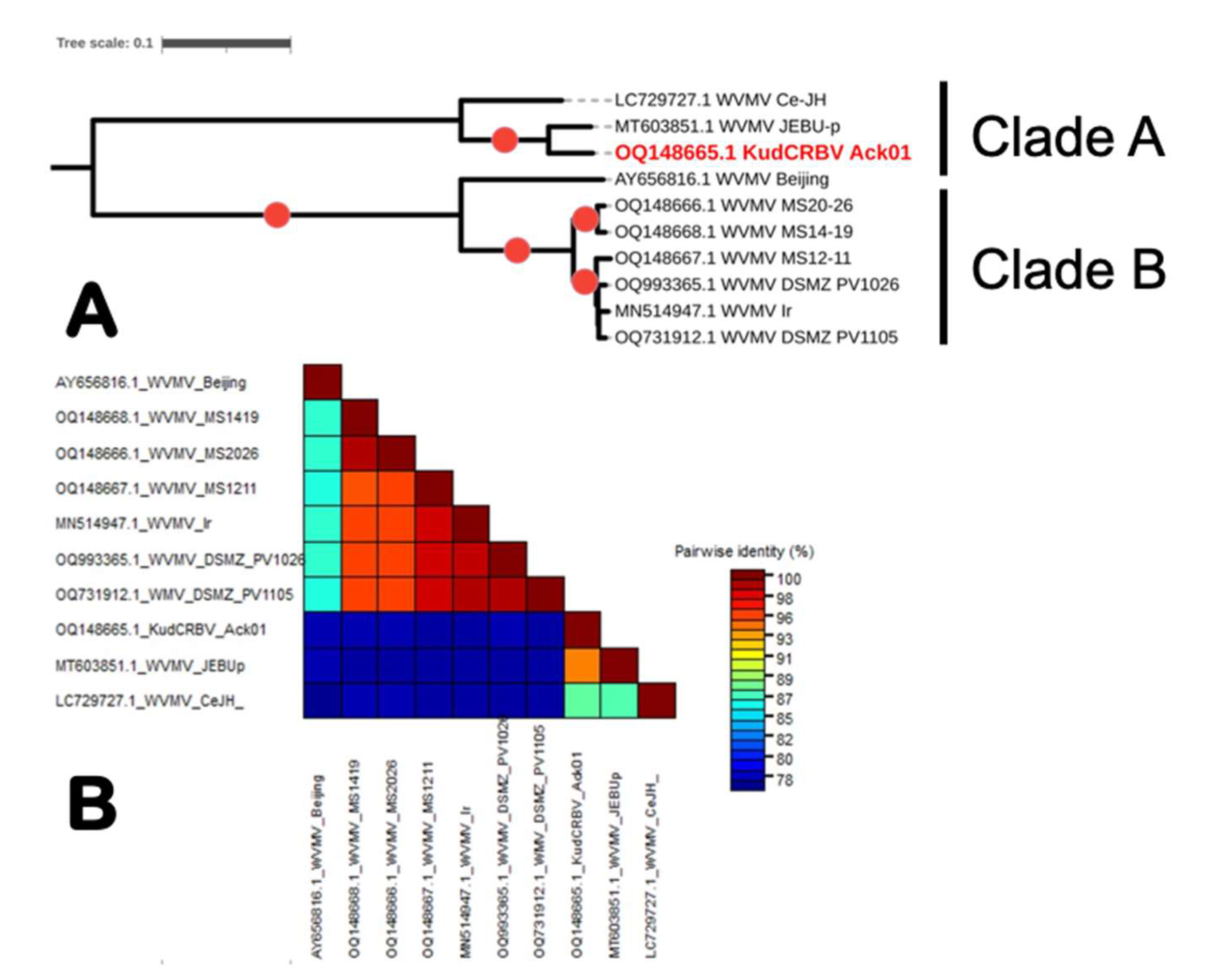
3.3. Genome Sequence Analyses of the Three Isolates of Wisteria Vein Mosaic Virus from Mississippi
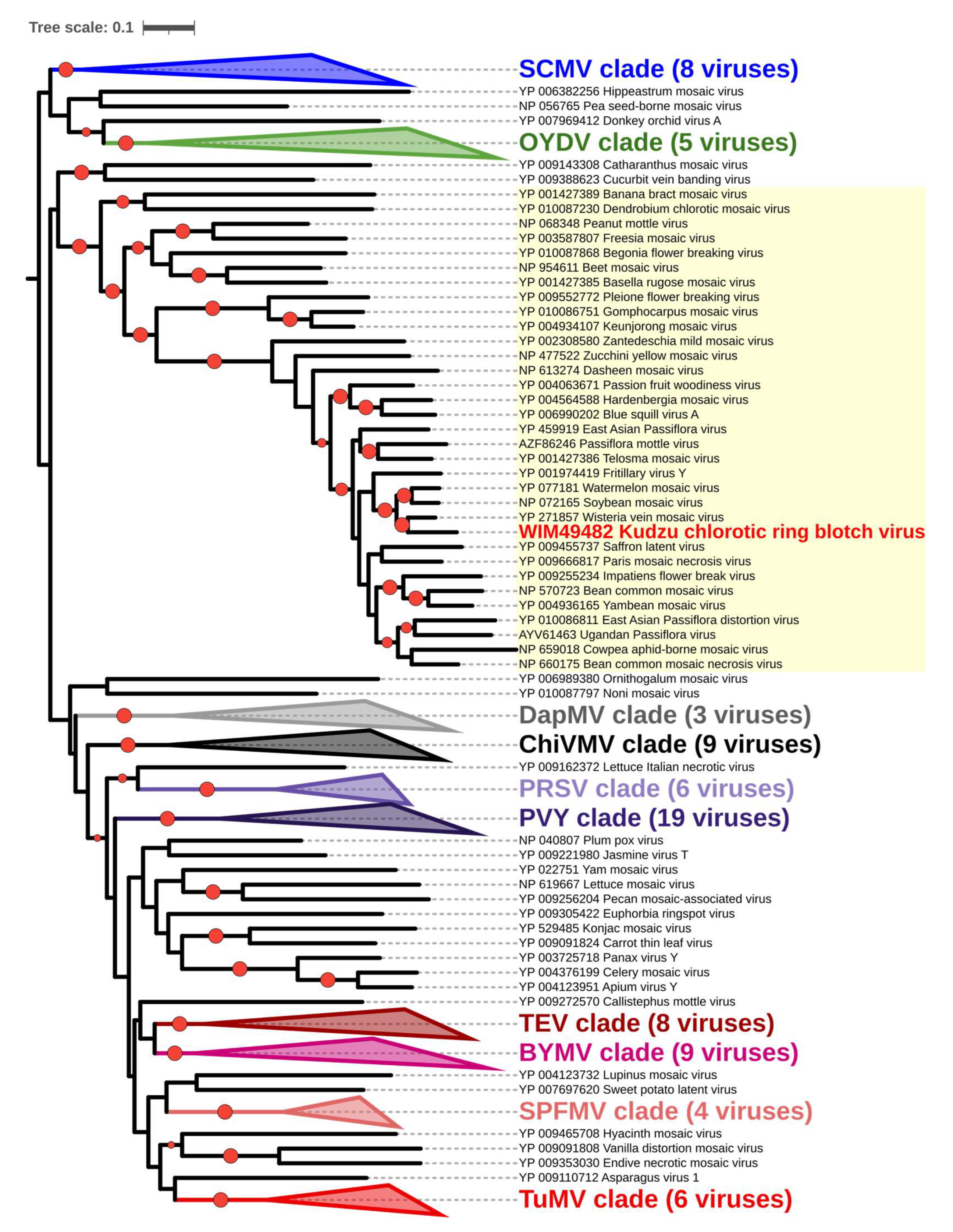
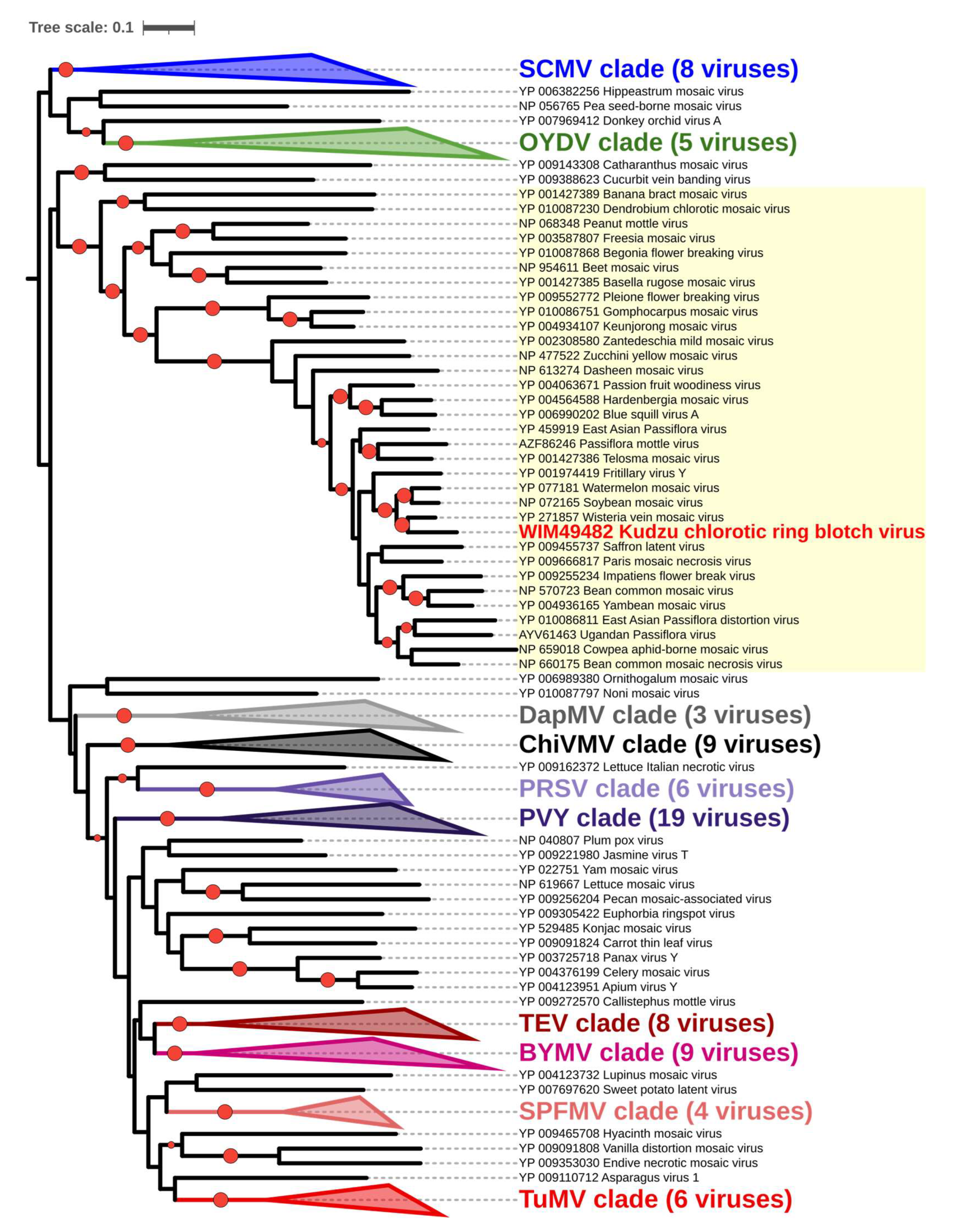
3.4. Comparison of KudCRBV with Allied Viruses
3.5. Survey and Transmission Experiments
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blaustein, R.J. Kudzu’s Invasion into Southern United States Life and Culture. In The Great Reshuffling: Human Dimensions of Invasive Alien Species; McNeely, J.A., Ed.; IUCN: Gland, Switzerland; Cambridge, UK, 2001; pp. 55–62. [Google Scholar]
- Forseth, I.N., Jr.; Innis, A.F. Kudzu (Pueraria montana): History, physiology, and ecology combine to make a major ecosystem threat. CRC Crit. Rev. Plant. Sci. 2004, 23, 401–413. [Google Scholar] [CrossRef]
- Sikora, E. Kudzu: Invasive weed supports the soybean rust pathogen through winter months in southeastern United States. Outlooks Pest Manag. 2014, 25, 175–179. [Google Scholar] [CrossRef]
- Harron, P.; Joshi, O.; Edgar, C.B.; Paudel, S.; Adhikari, A. Predicting kudzu (Pueraria montana) spread and its economic impacts in timber industry: A case study from Oklahoma. PLoS ONE 2020, 15, e0229835. [Google Scholar] [CrossRef] [PubMed]
- Bonde, M.R.; Nester, S.E.; Moore, W.F.; Allen, T.W. Comparative susceptibility of kudzu accessions from the southeastern United States to infection by Phakopsora pachyrhizi. Plant Dis. 2009, 93, 593–598. [Google Scholar] [CrossRef]
- Aboughanem-Sabanadzovic, N.; Allen, T.W.; Broome, M.; Lawrence, A.; Moore, W.F.; Sabanadzovic, S. First report of kudzu (Pueraria montana) infections by tobacco ringspot virus in Mississippi. Plant Dis. 2014, 98, 1746. [Google Scholar] [CrossRef]
- Khankhum, S.; Bollich, P.; Valverde, R.A. First report of tobacco ringspot virus infecting kudzu (Pueraria montana) in Louisiana. Plant Dis. 2013, 97, 561. [Google Scholar] [CrossRef]
- Zhou, J.; Aboughanem-Sabanadzovic, N.; Sabanadzovic, S.; Tzanetakis, I.E. First report of soybean vein necrosis virus infecting kudzu (Pueraria montana) in the United States of America. Plant Dis. 2018, 102, 1674. [Google Scholar] [CrossRef]
- Ha, C.; Coombs, S.; Revill, R.; Harding, R.; Vu, M.; Dale, J. Molecular characterization of begomoviruses and DNA satellites from Vietnam: Additional evidence that the New World geminiviruses were present in the Old World prior to continental separation. J. Gen. Virol. 2008, 89, 312–326. [Google Scholar] [CrossRef]
- Liang, X.; Mei, S.; Yu, H.; Zhang, S.; Wu, J.; Cao, M. Mixed infection of an emaravirus, a crinivirus, and a begomovirus in Pueraria lobata (Willd) Ohwi. Front. Microbiol. 2022, 13, 926724. Available online: https://www.frontiersin.org/articles/10.3389/fmicb.2022.926724 (accessed on 29 August 2023). [CrossRef]
- Inoue-Nagata, A.K.; Jordan, R.; Kreuze, J.; Li, F.; López-Moya, J.J.; Mäkinen, K.; Ohshima, K.; Wylie, S.J. ICTV Virus Taxonomy Profile: Potyviridae 2022. J. Gen. Virol. 2022, 103, 001738. [Google Scholar] [CrossRef]
- Zerbini, F.M.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Alfenas-Zerbini, P.; Dempsey, D.M.; Dutilh, B.E.; García, M.L.; Hendrickson, R.C.; et al. Changes to virus taxonomy and the ICTV Statutes ratified by the International Committee on Taxonomy of Viruses (2023). Arch. Virol. 2023, 168, 175. [Google Scholar] [CrossRef] [PubMed]
- Valverde, R.A.; Nameth, S.T.; Jordan, R.L. Analysis of double-stranded RNA for plant virus diagnosis. Plant Dis. 1990, 74, 255–258. [Google Scholar]
- Abou-Ghanem, N.; Sabanadzovic, S.; Minafra, A.; Saldarelli, P.; Martelli, G.P. Some properties of grapevine leafroll-associated virus 2 and molecular organization of the 3’ region of the viral genome. J. Plant Pathol. 1998, 80, 37–46. Available online: https://www.jstor.org/stable/41997899 (accessed on 29 August 2023).
- Okada, R.; Kiyota, E.; Sabanadzovic, S.; Moriyama, H.; Fukuhara, T.; Saha, P.; Roossinck, M.J.; Severin, A.; Valverde, R.A. Bell pepper endornavirus: Molecular and biological properties and occurrence in the genus Capsicum. J. Gen. Virol. 2011, 92, 2664–2673. [Google Scholar] [CrossRef] [PubMed]
- Valverde, R.A.; Sabanadzovic, S. A new plant virus with unique properties infecting Japanese holly fern. J. Gen. Virol. 2009, 90, 2542–2549. [Google Scholar] [CrossRef] [PubMed]
- Martelli, G.P.; Russo, M. Use of thin sectioning for the visualization and identification of plant viruses. Methods Virol. 1984, 8, 143–224. [Google Scholar]
- Froussard, P. A random-PCR method (rPCR) to construct whole cDNA library from low amounts of RNA. Nucleic Acids Res. 1992, 20, 2900. [Google Scholar] [CrossRef] [PubMed]
- Sabanadzovic, S.; Abou Ghanem-Sabanadzovic, N.; Gorbalenya, A.E. Permutation of the active site of putative RNA-dependent RNA polymerase in a newly identified species of plant alpha-like virus. Virology 2009, 394, 1–7. [Google Scholar] [CrossRef]
- Muhire, B.M.; Varsani, A.; Martin, D.P. SDT: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Capella-Gutierez, S.; Silla-Martinez, J.M.; Gabaldòn, T. TrimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucl. Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Blackman, R.L.; Eastop, V.F. Aphids on the World’s Crops: An Identification and Information Guide, 2nd ed.; John Wiley & Sons Ltd.: Chichester, UK, 2000; p. 414. ISBN 978-0-471-85191-2. [Google Scholar]
- Susko, D.J.; Mueller, J.P.; Spears, J.F. An evaluation of methods for breaking seed dormancy in kudzu (Pueraria lobata). Can. J. Bot. 2001, 79, 197–203. [Google Scholar] [CrossRef]
- Chung, B.Y.-W.; Allen Miller, W.; Atkins, J.F.; Firth, A.F. An overlapping essential gene in the Potyviridae. Proc. Natl. Acad. Sci. USA 2008, 105, 5897–5902. [Google Scholar] [CrossRef] [PubMed]
- Vijayapalani, P.; Maeshima, M.; Nagasaki-Takekuchi, N.; Miller, W.A. Interaction of the trans-frame potyvirus protein P3N-PIPO with host protein PCaP1 facilitates potyvirus movement. PLoS Pathog. 2012, 8, e1002639. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.X.; Song, L.M.; Tian, G.Z.; Li, X.F.; Fan, Z.F. The genomic sequence of wisteria vein mosaic virus and its similarities with other potyviruses. Arch. Virol. 2006, 151, 2311–2319. [Google Scholar] [CrossRef] [PubMed]
- Al-Jaberi, M.S.; Moradi, Z.; Mehrvar, M.; Al-Inizi, H.R.; Zakiaghl, M. Whole genome characterization of wisteria vein mosaic virus from Iran and its relationship to other members of bean common mosaic virus group. 3 Biotech 2021, 11, 407. [Google Scholar] [CrossRef]
- Jo, Y.; Yoon, Y.N.; Jang, Y.-W.; Choi, H.; Lee, Y.-H.; Kim, S.-M.; Choi, S.Y.; Lee, B.C.; Cho, W.K. Soybean viromes in the Republic of Korea revealed by RT-PCR and next-generation sequencing. Microorganisms 2020, 8, 1777. Available online: https://www.mdpi.com/2076-2607/8/11/1777 (accessed on 29 August 2023). [CrossRef] [PubMed]
- Naidu, R.A.; Karthikeyan, G. First report of Wisteria vein mosaic virus in Wisteria sinensis in the United States of America. Plant Health Prog. 2008, 9, 42. [Google Scholar] [CrossRef]
- Allen, T.; Hollier, C.; Sikora, E. A continuing saga: Soybean rust in the continental United States, 2004 to 2013. Outlooks Pest Manag. 2014, 25, 167–174. [Google Scholar] [CrossRef]
- Roossinck, M.J.; García-Arenal, F. Ecosystem simplification, biodiversity loss and plant virus emergence. Curr. Opin. Virol. 2015, 10, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.H.; Whitham, S.A. Control of virus diseases in soybeans. Adv. Virus Res. 2014, 90, 355–390. [Google Scholar] [CrossRef] [PubMed]
- Shukla, D.D.; Ward, C.W. Structure of potyvirus coat proteins and its application in the taxonomy of the potyvirus group. Adv. Virus Res. 1989, 36, 273–314. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.J.; Antoniw, J.F.; Fauquet, C.M. Molecular criteria for genus and species discrimination within the family Potyviridae. Arch Virol. 2005, 150, 459–479. [Google Scholar] [CrossRef]
- Gibbs, A.J.; Hajizadeh, M.; Ohshima, K.; Jones, R.A. The potyviruses: An evolutionary synthesis is emerging. Viruses 2020, 12, 132. [Google Scholar] [CrossRef]
- Gibbs, A.J.; Ohshima, K.; Phillips, M.J.; Gibbs, M.J. The prehistory of potyviruses: Their initial radiation was during the dawn of agriculture. PLoS ONE 2008, 3, e2523. [Google Scholar] [CrossRef]
- Gibbs, A.J.; Trueman, J.; Gibbs, M.J. The bean common mosaic virus lineage of potyviruses: Where did it arise and when? Arch. Virol. 2008, 153, 2177–2187. [Google Scholar] [CrossRef]
- Brierley, P.; Lorentz, P. Wisteria mosaic and peony leaf curl, two diseases of ornamental plants caused by viruses transmissible by grafting but not by sap inoculation. Plant Dis. Reptr. 1957, 41, 691–693. [Google Scholar]
- Bos, L. The identification of three new viruses isolated from Wisteria and Pisum in The Netherlands, and the problem of variation within the potato virus Y group. Neth. J. Plant Pathol. 1970, 76, 8–46. [Google Scholar] [CrossRef]
- Clover, G.; Denton, J.; Denton, G. First report of wisteria vein mosaic virus on Wisteria spp. in the United Kingdom. New Dis. Rep. 2015, 31, 2044-0588. [Google Scholar] [CrossRef]
- Kamińska, M.; Malinowski, T.; Rudzińska-Langwald, A.; Diaz, L. The occurrence of wisteria vein mosaic virus in Wisteria floribunda DC plants in Poland. J. Phytopathol. 2006, 154, 414–417. [Google Scholar] [CrossRef]
- D’Attoma, G.; Minafra, A.; Saldarelli, P.; Morelli, M. Molecular evidence for the presence of wisteria vein mosaic virus in Italy: Shedding light on genetic diversity and evolutionary dynamics of virus geographic populations. Agriculture 2023, 13, 1090. [Google Scholar] [CrossRef]
- Ward, L.; Tang, J.; Clover, G. First report of wisteria vein mosaic virus on Wisteria sinensis in New Zealand. Plant Dis. 2008, 92, 1134. [Google Scholar] [CrossRef] [PubMed]
- Al-Jaberi, M.; Mehrvar, M.; Zakiahgl, M. Molecular identification of an isolate of wisteria vein mosaic virus in Khorasan Razavi province. J. Plant Prot. 2019, 33, 23–26. Available online: https://jpp.um.ac.ir/article_37452.html (accessed on 3 September 2023). (In Persian).
- Valouzi, H.; Hashemi, S.-S.; Wylie, S.J.; Ahadiyat, A.; Golnaraghi, A. Wisteria vein mosaic virus detected for the first time in Iran from an unknown host by analysis of aphid vectors. Plant Pathol. J. 2020, 36, 87–97. [Google Scholar] [CrossRef]
- Ji, Z.-L.; Zhu, P.-X.; Ji, Y.-H.; Xu, F.; Zhu, F. First report of wisteria vein mosaic virus in Chinese wisteria in Jiangxi Province in China. J. Plant Pathol. 2019, 101, 1259–1260. [Google Scholar] [CrossRef]
- Zhu, F.; Zhu, P.-X.; Xu, F.; Ji, Z.-L. First report of wisteria vein mosaic virus infecting Chinese wisteria in Jiangsu Province in China. J. Plant Dis. Prot. 2019, 126, 373–377. [Google Scholar] [CrossRef]
- Choi, H.; Jo, Y.; Chung, H.; Choi, S.Y.; Kim, S.-M.; Hong, J.-S.; Lee, B.C.; Cho, W.K. Phylogenetic and phylodynamic analyses of soybean mosaic virus using 305 coat protein gene sequences. Plants 2022, 11, 3256. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aboughanem-Sabanadzovic, N.; Stephenson, R.C.; Allen, T.W.; Henn, A.; Moore, W.F.; Lawrence, A.; Sabanadzovic, S. Characterization of a Putative New Member of the Genus Potyvirus from Kudzu (Pueraria montana var. lobata) in Mississippi. Viruses 2023, 15, 2145. https://doi.org/10.3390/v15112145
Aboughanem-Sabanadzovic N, Stephenson RC, Allen TW, Henn A, Moore WF, Lawrence A, Sabanadzovic S. Characterization of a Putative New Member of the Genus Potyvirus from Kudzu (Pueraria montana var. lobata) in Mississippi. Viruses. 2023; 15(11):2145. https://doi.org/10.3390/v15112145
Chicago/Turabian StyleAboughanem-Sabanadzovic, Nina, Ronald Christian Stephenson, Thomas W. Allen, Alan Henn, William F. Moore, Amanda Lawrence, and Sead Sabanadzovic. 2023. "Characterization of a Putative New Member of the Genus Potyvirus from Kudzu (Pueraria montana var. lobata) in Mississippi" Viruses 15, no. 11: 2145. https://doi.org/10.3390/v15112145
APA StyleAboughanem-Sabanadzovic, N., Stephenson, R. C., Allen, T. W., Henn, A., Moore, W. F., Lawrence, A., & Sabanadzovic, S. (2023). Characterization of a Putative New Member of the Genus Potyvirus from Kudzu (Pueraria montana var. lobata) in Mississippi. Viruses, 15(11), 2145. https://doi.org/10.3390/v15112145