
Genomic Analysis of Two Cold-Active Pseudoalteromonas Phages Isolated from the Continental Shelf in the Arctic Ocean

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Area and Sample Collection
2.2. Isolation and Identification of Bacterial Strains
2.3. Isolation of Phages
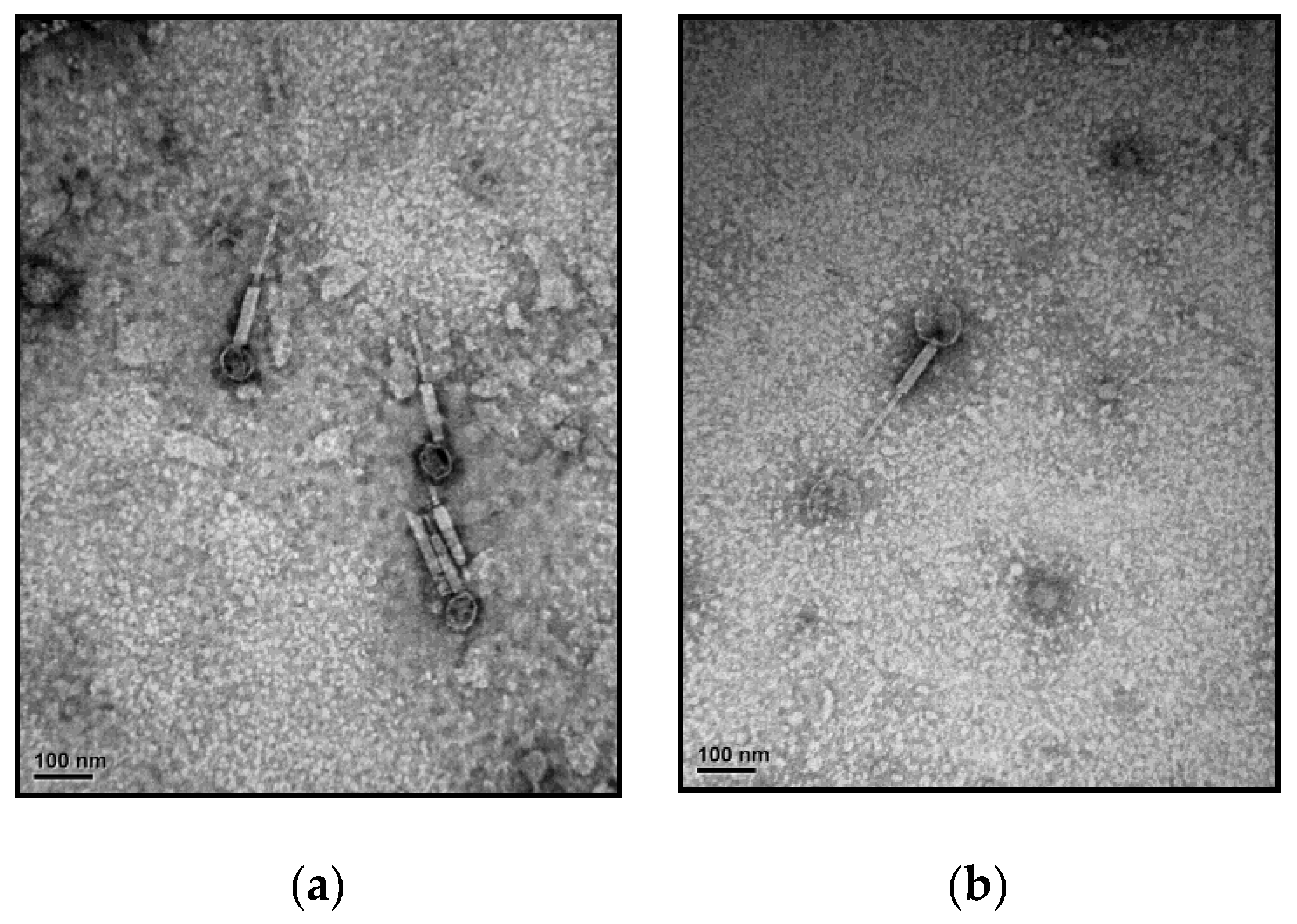
2.4. Physiological and Morphological Characterization
2.5. Concentration and Purification of Phages
2.6. Genome Sequencing of Phages and Host Strains
2.7. Genome Annotation
2.8. Taxonomic Analysis
2.9. P2-like Database
2.10. Modular Divergence
2.11. Timetree Analysis
2.12. Mass Spectrometry
2.13. Nucleotide Sequence Accession Numbers
3. Results
3.1. Isolation and Characterization of the Phage–Host Systems
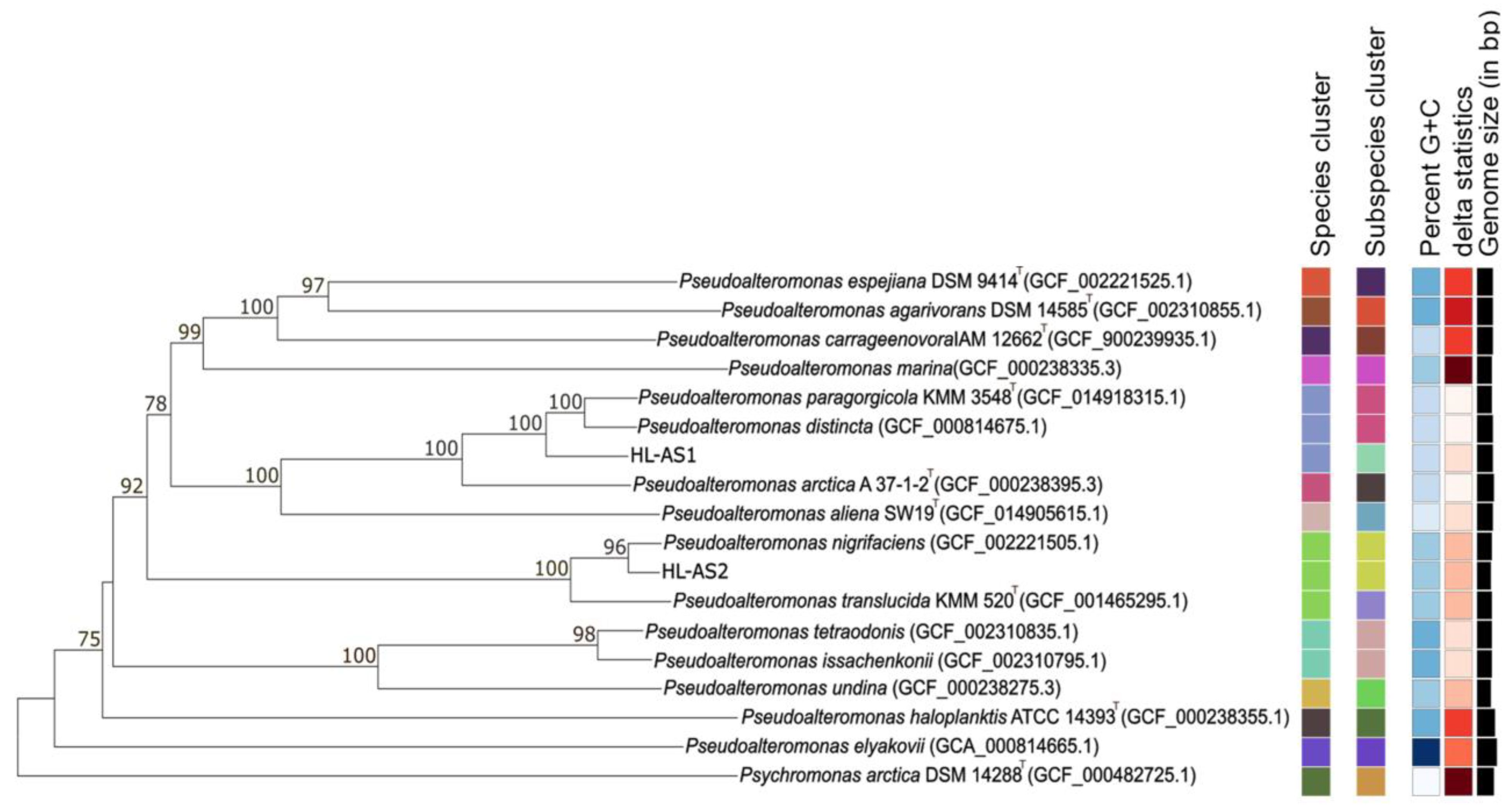
3.2. Taxonomic Classification of Host Strains
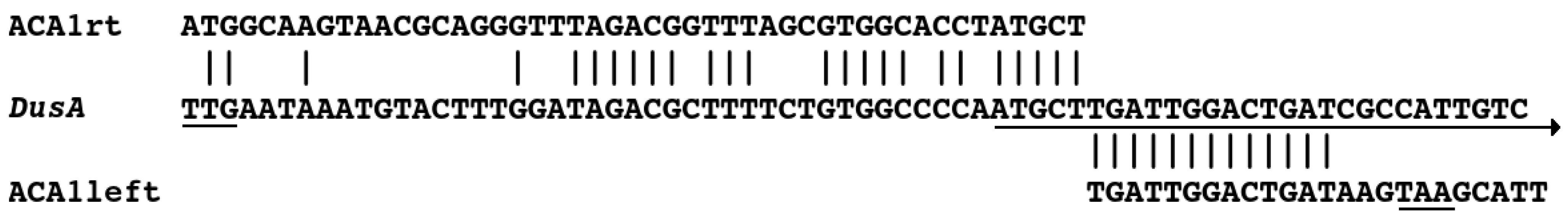
3.3. Sequencing and Relationship to Prophage proACA1-A
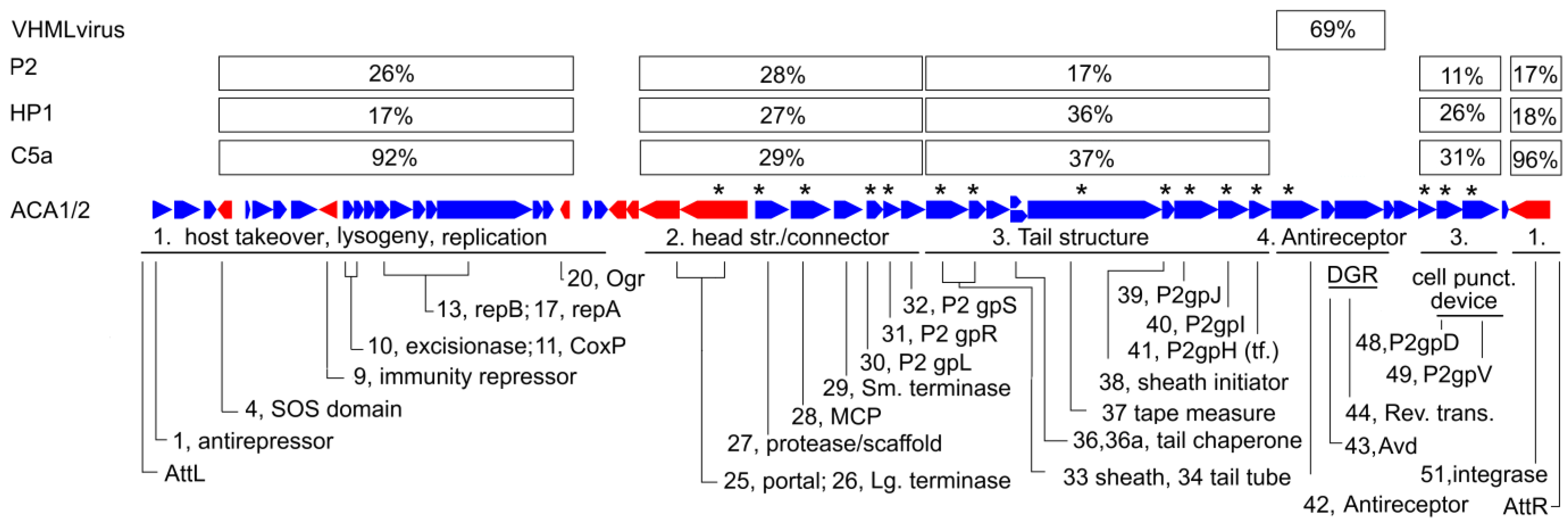
3.4. Genome Annotation
3.5. General Features of the Genome
3.6. Head Structure and Connector Module
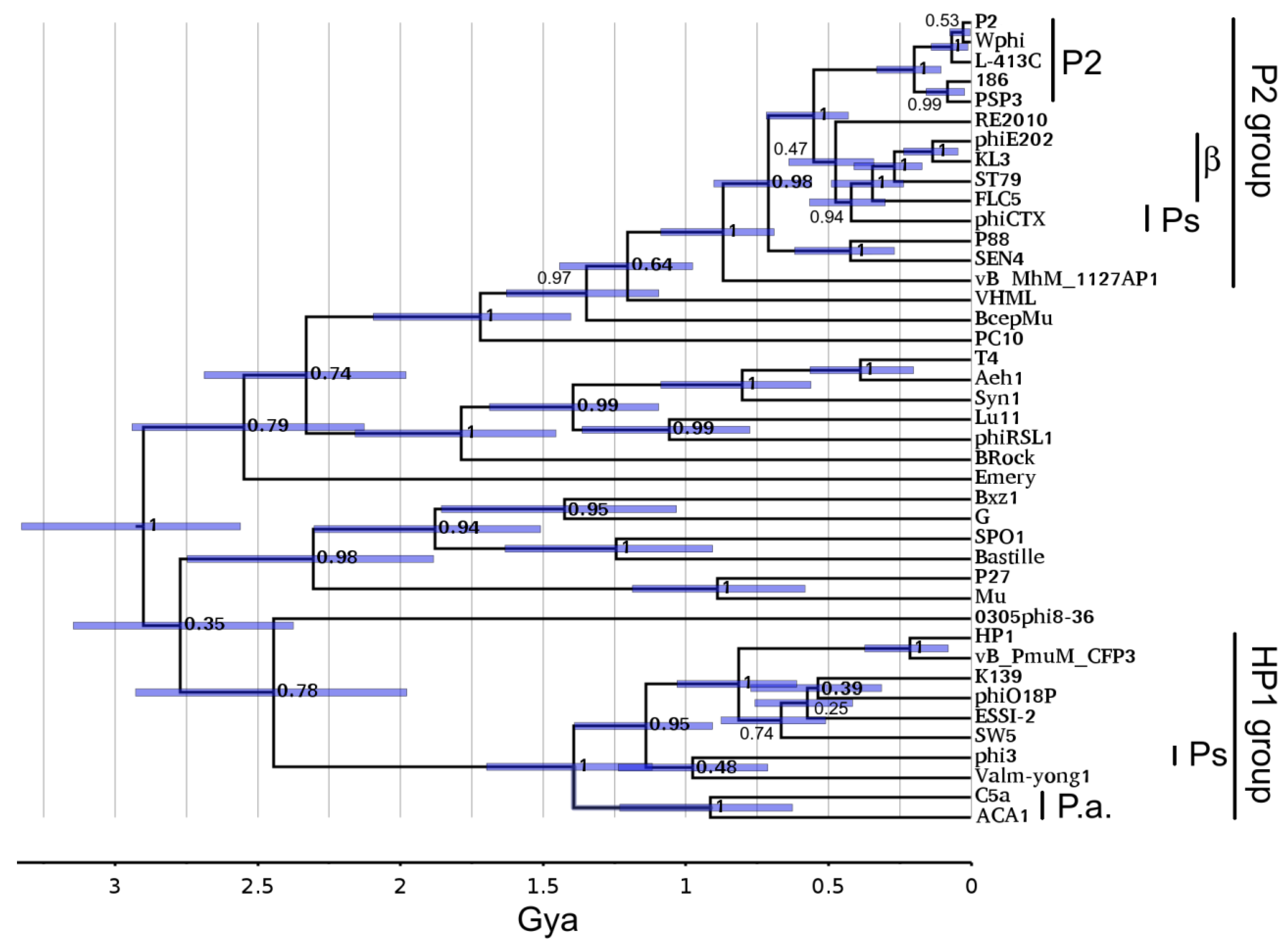
3.7. Portal Timetree
3.8. Time of Residence of Phage Lineages in Pseudoalteromonas
3.9. Nonstructural Module
3.10. The C5a Connection
3.11. Tail Structure Module
3.12. Sheath Timetree
3.13. Antirepressor and Diversity Generating Retroelement
4. Discussion
4.1. Classification
4.2. History of the P2-Like Phages
4.3. The DGR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Koskella, B.; Meaden, S. Understanding bacteriophage specificity in natural microbial communities. Viruses 2013, 5, 806–823. [Google Scholar] [CrossRef]
- Wells, L.E. Cold-active viruses. In Psychrophiles: From Biodiversity to Biotechnology; Wells, L.E., Ed.; Springer: New York, NY, USA, 2008; pp. 157–173. [Google Scholar] [CrossRef]
- Wells, L.E.; Deming, J.W. Characterization of a cold-active bacteriophage on two psychrophilic marine hosts. Aquat. Microb. Ecol. 2006, 45, 15–29. [Google Scholar] [CrossRef]
- Steward, G.; Smith, D.; Azam, F. Abundance and production of bacteria and viruses in the Bering and Chukchi Seas. Mar. Ecol. Prog. Ser. 1996, 131, 287–300. [Google Scholar] [CrossRef]
- Danovaro, R.; Corinaldesi, C.; Dell’anno, A.; Fuhrman, J.A.; Middelburg, J.J.; Noble, R.T.; Suttle, C.A. Marine viruses and global climate change. FEMS Microbiol. Rev. 2011, 35, 993–1034. [Google Scholar] [CrossRef]
- Luhtanen, A.-M.; Eronen-Rasimus, E.; Oksanen, H.M.; Tison, J.-L.; Delille, B.; Dieckmann, G.S.; Rintala, J.-M.; Bamford, D.H. The first known virus isolates from Antarctic sea ice have complex infection patterns. FEMS Microbiol. Ecol. 2018, 94, fiy028. [Google Scholar] [CrossRef]
- Demina, T.A.; Luhtanen, A.-M.; Roux, S.; Oksanen, H.M. Virus-host interactions and genetic diversity of Antarctic sea ice bacteriophages. mBio 2022, 13, e00651-22. [Google Scholar] [CrossRef]
- Yu, Z.-C.; Chen, X.-L.; Shen, Q.-T.; Zhao, D.-L.; Tang, B.-L.; Su, H.-N.; Wu, Z.-Y.; Qin, Q.-L.; Xie, B.-B.; Zhang, X.-Y.; et al. Filamentous phages prevalent in Pseudoalteromonas Spp. confer properties advantageous to host survival in Arctic sea ice. ISME J. 2015, 9, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C.Y.; Lee, I.; Hwang, Y.J.; Yoon, S.J.; Lee, W.S.; Cho, B.C. Pseudoalteromonas neustonica sp. nov., isolated from the sea surface microlayer of the Ross Sea (Antarctica), and emended description of the genus Pseudoalteromonas. Int. J. Syst. Evol. Microbiol. 2016, 66, 3377–3382. [Google Scholar] [CrossRef] [PubMed]
- Bowman, J.P.; McMeekin, T.A. Pseudoalteromonas. In Bergey’s Manual of Systematics of Archaea and Bacteria; John Wiley & Sons: Hoboken, NJ, USA, 2015; pp. 1–22. [Google Scholar] [CrossRef]
- Bosi, E.; Fondi, M.; Orlandini, V.; Perrin, E.; Maida, I.; de Pascale, D.; Tutino, M.L.; Parrilli, E.; Lo Giudice, A.; Filloux, A.; et al. The pangenome of (Antarctic) Pseudoalteromonas bacteria: Evolutionary and functional insights. BMC Genom. 2017, 18, 93. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.; Evans, F.F.; Schleheck, D.; Mai-Prochnow, A.; Burke, C.; Penesyan, A.; Dalisay, D.S.; Stelzer-Braid, S.; Saunders, N.; Johnson, J.; et al. Analysis of the Pseudoalteromonas tunicata genome reveals properties of a surface-associated life style in the marine environment. PLoS ONE 2008, 3, e3252. [Google Scholar] [CrossRef] [PubMed]
- Hardies, S.C.; Hwang, Y.J.; Hwang, C.Y.; Jang, G.I.; Cho, B.C. Morphology, physiological characteristics, and complete sequence of marine bacteriophage ϕRIO-1 infecting Pseudoalteromonas marina. J. Virol. 2013, 87, 9189–9198. [Google Scholar] [CrossRef]
- Middelboe, M.; Nielsen, T.G.; Bjørnsen, P.K. Viral and bacterial production in the North Water: In situ measurements, batch-culture experiments and characterization and distribution of a virus–host system. Deep Sea Res. Part II Top. Stud. Oceanogr. 2002, 49, 5063–5079. [Google Scholar] [CrossRef]
- Zheng, K.; Dong, Y.; Liang, Y.; Liu, Y.; Zhang, X.; Zhang, W.; Wang, Z.; Shao, H.; Sung, Y.Y.; Mok, W.J.; et al. Genomic diversity and ecological distribution of marine Pseudoalteromonas phages. Mar. Life Sci. Technol. 2023, 5, 271–285. [Google Scholar] [CrossRef]
- Christie, G.E.; Calendar, R. Bacteriophage P2. Bacteriophage 2016, 6, e1145782. [Google Scholar] [CrossRef]
- Nilsson, A.S.; Haggård-Ljungquist, E. Evolution of P2-like Phages and their impact on bacterial evolution. Res. Microbiol. 2007, 158, 311–317. [Google Scholar] [CrossRef]
- Nilsson, A.S.; Ljungquist, E.H. The P2-Like Bacteriophages. In The Bacteriophages, 2nd ed.; Abedon, S.T., Calendar, R.L., Eds.; Oxford University Press: Oxford, UK, 2006; pp. 365–390. [Google Scholar]
- Lavigne, R.; Darius, P.; Summer, E.; Seto, D.; Mahadevan, P.; Nilsson, A.; Ackermann, H.; Kropinski, A. Classification of Myoviridae bacteriophages using protein sequence similarity. BMC Microbiol. 2009, 9, 224. [Google Scholar] [CrossRef]
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Alfenas-Zerbini, P.; Dempsey, D.M.; Dutilh, B.E.; García, M.L.; Hendrickson, R.C.; et al. Recent changes to virus taxonomy ratified by the international committee on taxonomy of viruses. Arch. Virol. 2022, 167, 2429–2440. [Google Scholar] [CrossRef]
- Hwang, C.Y.; Cho, B.C. Cohaesibacter gelatinilyticus gen. nov., sp. nov., a marine bacterium that forms a distinct branch in the order Rhizobiales, and proposal of Cohaesibacteraceae fam. nov. Int. J. Syst. Evol. Microbiol. 2008, 58, 267–277. [Google Scholar] [CrossRef]
- Yoon, S.-H.; Ha, S.-M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef]
- Van Twest, R.; Kropinski, A.M. Bacteriophage enrichment from water and soil. In Bacteriophages: Methods and Protocols; Van Twest, R., Kropinski, A.M., Eds.; Oxford University Press: Oxford, UK, 2009; Volume 1, pp. 15–21. ISBN 9781588296825. [Google Scholar] [CrossRef]
- Hwang, C.Y.; Cho, Y.; Jang, G.I.; Cho, B.C.; Hardies, S.C. Complete genome sequence of Sulfitobacter phage ΦGT1, isolated from a tidal flat. Microbiol. Resour. Announc. 2020, 9, e00779-20. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Holt, K.E. Assembling the perfect bacterial genome using Oxford Nanopore and Illumina sequencing. PLoS Comput. Biol. 2023, 19, e1010905. [Google Scholar] [CrossRef]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Thomas, J.A.; Rolando, M.R.; Carroll, C.A.; Shen, P.S.; Belnap, D.M.; Weintraub, S.T.; Serwer, P.; Hardies, S.C. Characterization of Pseudomonas chlororaphis myovirus 201ϕ2-1 via genomic sequencing, mass spectrometry, and electron microscopy. J. Virol. 2008, 376, 330–338. [Google Scholar] [CrossRef]
- Zimmermann, L.; Stephens, A.; Nam, S.-Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A Completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef]
- Gabler, F.; Nam, S.-Z.; Till, S.; Mirdita, M.; Steinegger, M.; Söding, J.; Lupas, A.N.; Alva, V. Protein sequence analysis using the MPI Bioinformatics Toolkit. Curr. Protoc. Bioinformatics 2020, 72, e108. [Google Scholar] [CrossRef]
- Consortium, T.U.; Bateman, A.; Martin, M.-J.; Orchard, S.; Magrane, M.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bye-A-Jee, H.; et al. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar] [CrossRef]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef]
- Nishimura, Y.; Yoshida, T.; Kuronishi, M.; Uehara, H.; Ogata, H.; Goto, S. ViPTree: The viral proteomic tree server. Bioinformatics 2017, 33, 2379–2380. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Göker, M. VICTOR: Genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Göker, M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 2019, 10, 2182. [Google Scholar] [CrossRef]
- Yoon, S.-H.; Ha, S.-M.; Lim, J.; Kwon, S.; Chun, J. A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Leeuwenhoek 2017, 110, 1281–1286. [Google Scholar] [CrossRef] [PubMed]
- Hardies, S.C.; Cho, B.C.; Jang, G.I.; Wang, Z.; Hwang, C.Y. Identification of structural and morphogenesis genes of Sulfitobacter phage ΦGT1 and placement within the evolutionary history of the Podoviruses. Viruses 2023, 15, 1475. [Google Scholar] [CrossRef] [PubMed]
- González, B.; Li, D.; Li, K.; Wright, E.T.; Hardies, S.C.; Thomas, J.A.; Serwer, P.; Jiang, W. Structural studies of the phage G tail demonstrate an atypical tail contraction. Viruses 2021, 13, 2094. [Google Scholar] [CrossRef] [PubMed]
- Swofford, D.L. PAUP: Phylogenetic Analysis Using Parsimony (and Other Methods); Sinauer Associates: Sunderland, UK, 2001. [Google Scholar]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Hoover, R.B.; Pikuta, E.V. Psychrophilic and psychrotolerant microbial extremophiles in polar environments. In Polar microbiology: The Ecology, Biodiversity and Bioremediation Potential of Microorganisms in Extremely Cold Environments; Bej, A.K., Aislabie, J., Atlas, R.M., Eds.; CRC Press Taylor & Francis Group: Oxfordshire, UK, 2010; pp. 115–156. [Google Scholar]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiolo. 2007, 57, 81–91. [Google Scholar] [CrossRef]
- Karlsson, J.L.; Cardoso-Palacios, C.; Nilsson, A.S.; Haggård -Ljungquist, E. Evolution of immunity and host chromosome integration site of P2-like coliphages. J. Bacteriol. 2006, 188, 3923–3935. [Google Scholar] [CrossRef]
- Chang, J.R.; Spilman, M.S.; Rodenburg, C.M.; Dokland, T. Functional domains of the bacteriophage P2 scaffolding protein: Identification of residues involved in assembly and protease activity. Virology 2009, 384, 144–150. [Google Scholar] [CrossRef]
- Hardies, S.C.; Thomas, J.A.; Black, L.; Weintraub, S.T.; Hwang, C.Y.; Cho, B.C. Identification of structural and morphogenesis genes of Pseudoalteromonas phage ΦRIO-1 and placement within the evolutionary history of Podoviridae. Virology 2016, 489, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Turner, D.; Kropinski, A.M.; Adriaenssens, E.M. A Roadmap for genome-based phage taxonomy. Viruses 2021, 13, 506. [Google Scholar] [CrossRef] [PubMed]
- Peat, T.S.; Frank, E.G.; McDonald, J.P.; Levine, A.S.; Woodgate, R.; Hendrickson, W.A. Structure of the UmuD′ protein and its regulation in response to DNA damage. Nature 1996, 380, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Shearwin, K.E.; Brumby, A.M.; Egan, J.B. The Tum protein of coliphage 186 is an antirepressor. J. Biol. Chem. 1998, 273, 5708–5715. [Google Scholar] [CrossRef] [PubMed]
- Oakey, H.J.; Cullen, B.R.; Owens, L. The complete nucleotide sequence of the Vibrio harveyi bacteriophage VHML. J. Appl. Microbiol. 2002, 93, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Summer, E.J.; Gonzalez, C.F.; Carlisle, T.; Mebane, L.M.; Cass, A.M.; Savva, C.G.; LiPuma, J.; Young, R. Burkholderia cenocepacia phage BcepMu and a family of Mu-like phages encoding potential pathogenesis factors. J. Mol. Biol. 2004, 340, 49–65. [Google Scholar] [CrossRef]
- Guo, H.; Arambula, D.; Ghosh, P.; Miller, J.F. Diversity-generating retroelements in phage and bacterial genomes. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phage | Host | Date | Sediment Core ID (Location) | Sediment Depth below Seafloor (m) | Bottom Water | ||
|---|---|---|---|---|---|---|---|
| Depth (m) | Temperature (℃) | Salinity | |||||
| ACA1 | HL-AS1 | 3 August 2011 | ARA02B/02-GC-01 (74.30° N, 167.65° W) | 1.9 | 322 | 0.7 | 34.8 |
| ACA2 | HL-AS2 | 2 August 2011 | ARA02B/01-GC-02 (73.64° N, 166.51° W) | 4.0 | 112 | −1.7 | 32.9 |
| HP1 Group | P2 Subgroup | |
|---|---|---|
| Bielevirus Catalunyavirus Hpunavirus Irrigatiovirus Irtavirus Longwoodvirus Phitrevirus Playavirus Seongnamvirus Valbvirus Vulnificusvirus Yongunavirus ACA phages (this study) | Aptresvirus Aresaunavirus Arsyunavirus Baylorvirus Bracchivirus Citexvirus Dagavirus Duodecimduovirus Eganvirus Elveevirus Entnonagintavirus Felsduovirus Gegavirus Gegevirus Gemsvirus | Kapieceevirus Kayeltresvirus Kisquattuordecimvirus Kisquinquevirus Nampongvirus Novemvirus Peduovirus Reginaelenavirus Reipivirus Senquatrovirus Simpcentumvirus Stockinghallvirus Tigrvirus Vimunumvirus Wadgaonvirus Xuanwuvirus |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hwang, C.Y.; Cho, B.C.; Kang, J.K.; Park, J.; Hardies, S.C. Genomic Analysis of Two Cold-Active Pseudoalteromonas Phages Isolated from the Continental Shelf in the Arctic Ocean. Viruses 2023, 15, 2061. https://doi.org/10.3390/v15102061
Hwang CY, Cho BC, Kang JK, Park J, Hardies SC. Genomic Analysis of Two Cold-Active Pseudoalteromonas Phages Isolated from the Continental Shelf in the Arctic Ocean. Viruses. 2023; 15(10):2061. https://doi.org/10.3390/v15102061
Chicago/Turabian StyleHwang, Chung Yeon, Byung Cheol Cho, Jin Kyeong Kang, Jihye Park, and Stephen C. Hardies. 2023. "Genomic Analysis of Two Cold-Active Pseudoalteromonas Phages Isolated from the Continental Shelf in the Arctic Ocean" Viruses 15, no. 10: 2061. https://doi.org/10.3390/v15102061
APA StyleHwang, C. Y., Cho, B. C., Kang, J. K., Park, J., & Hardies, S. C. (2023). Genomic Analysis of Two Cold-Active Pseudoalteromonas Phages Isolated from the Continental Shelf in the Arctic Ocean. Viruses, 15(10), 2061. https://doi.org/10.3390/v15102061