
Chetomin, a SARS-CoV-2 3C-like Protease (3CLpro) Inhibitor: In Silico Screening, Enzyme Docking, Molecular Dynamics and Pharmacokinetics Analysis

 , ,
, ,  , , ,
, , ,  ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. 3CLpro Preparation
2.2. Database Preparation
2.3. Molecular Docking
2.4. MD Simulations
2.5. Binding Affinity Computations
2.6. Physicochemical Features
2.7. Pharmacokinetic Characteristics
3. Results and Discussion
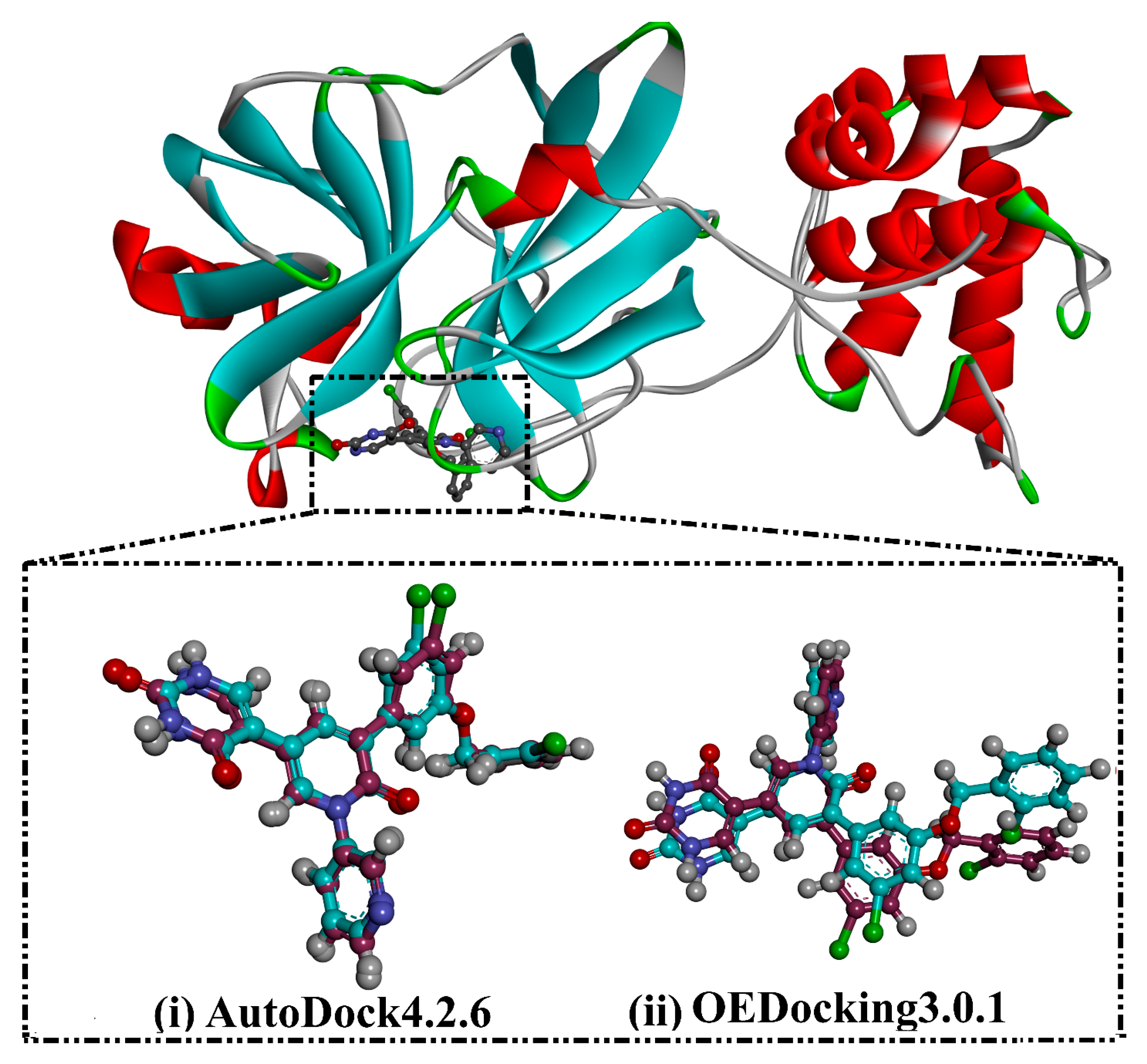
3.1. Docking Assessment
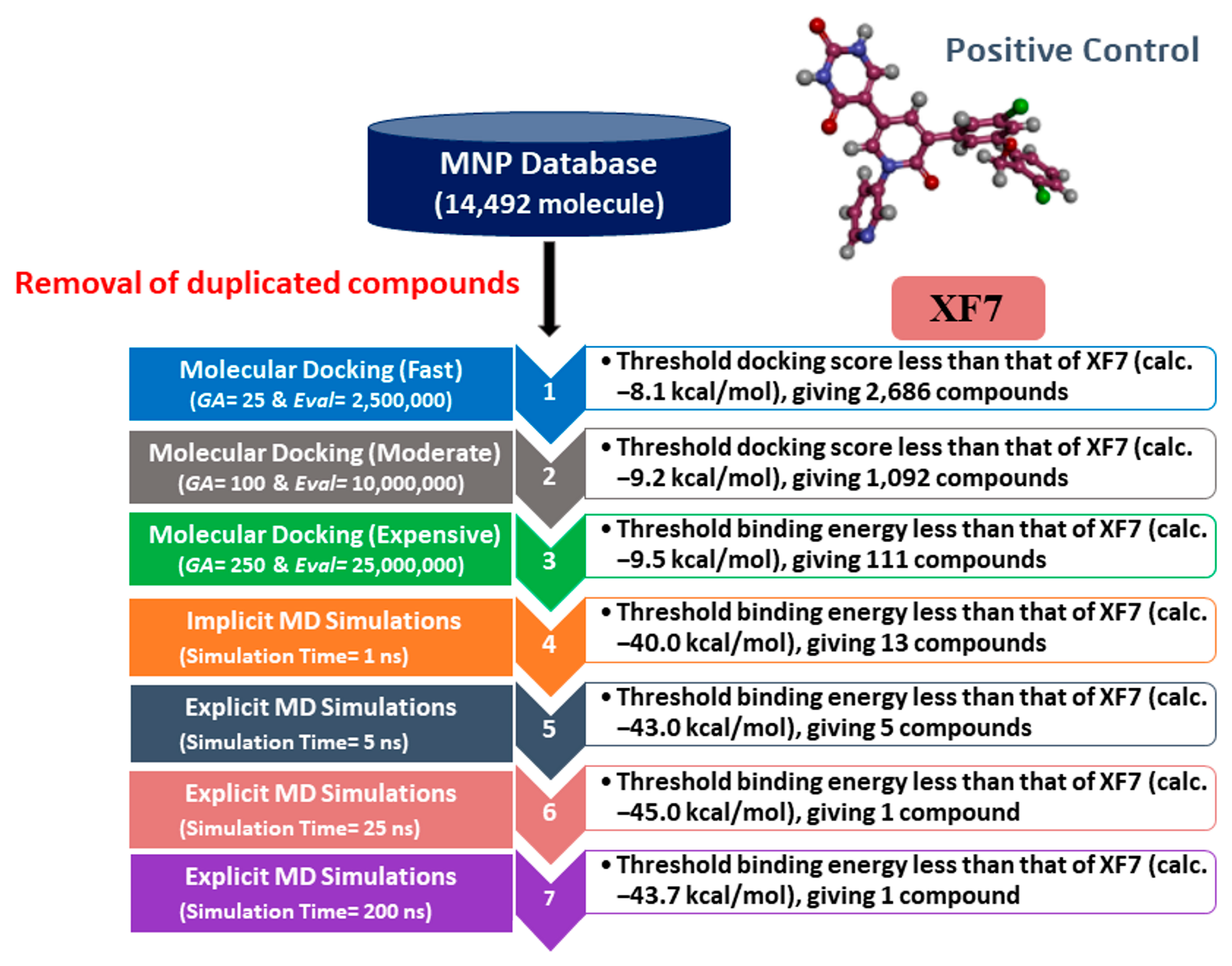
3.2. MNP Database Screening
3.3. MD Simulations
3.4. Post-MD Analyses
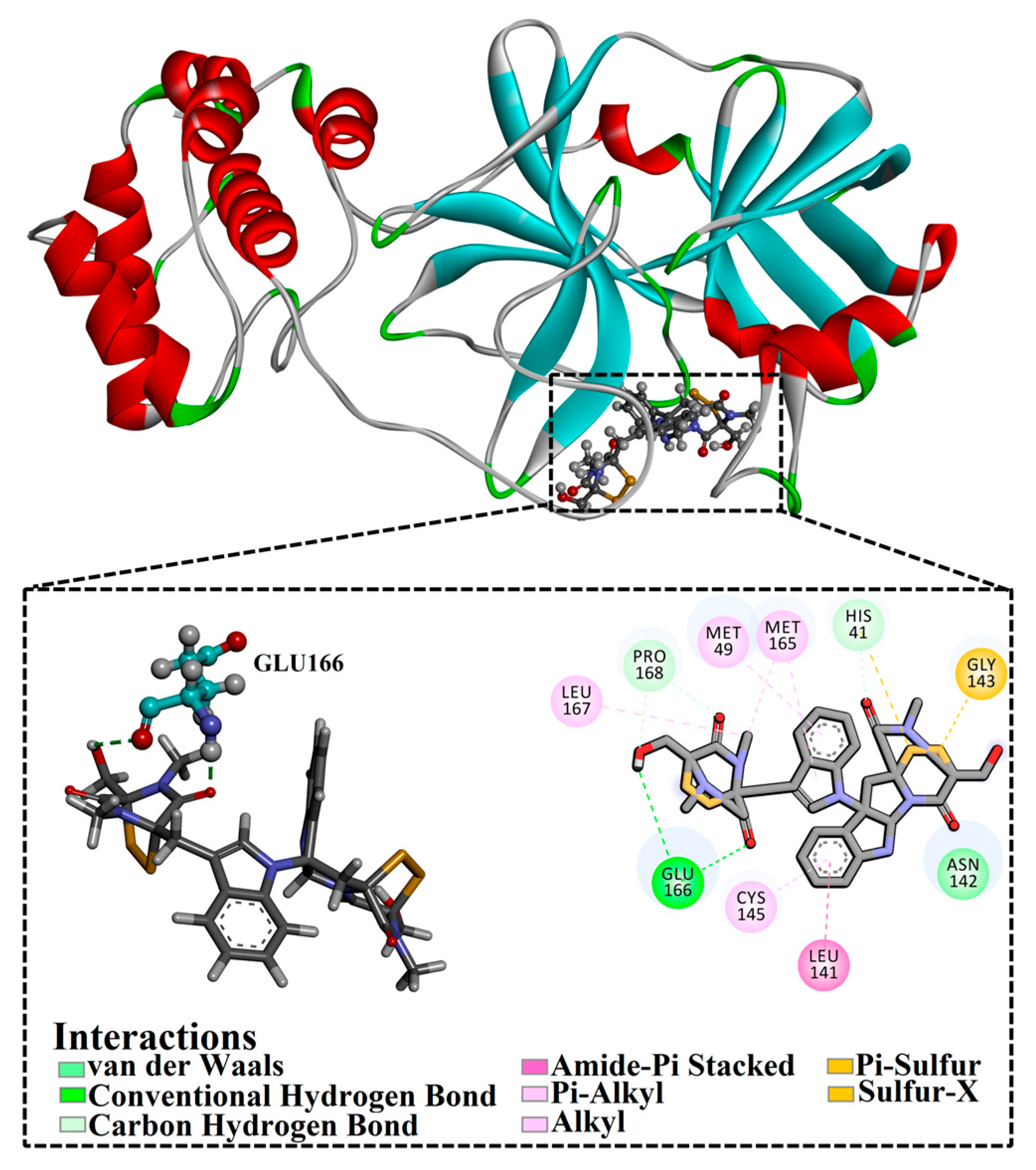
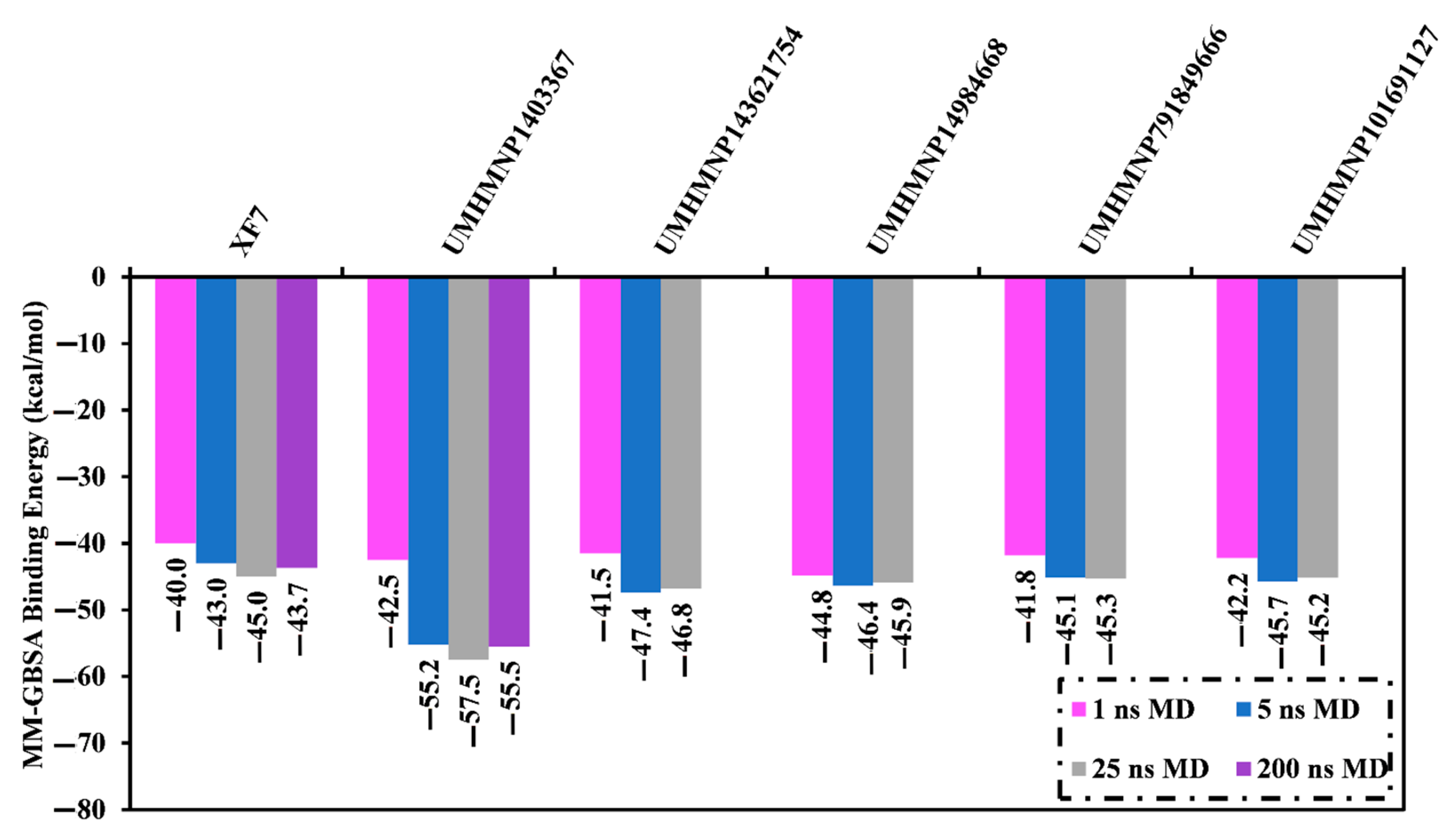
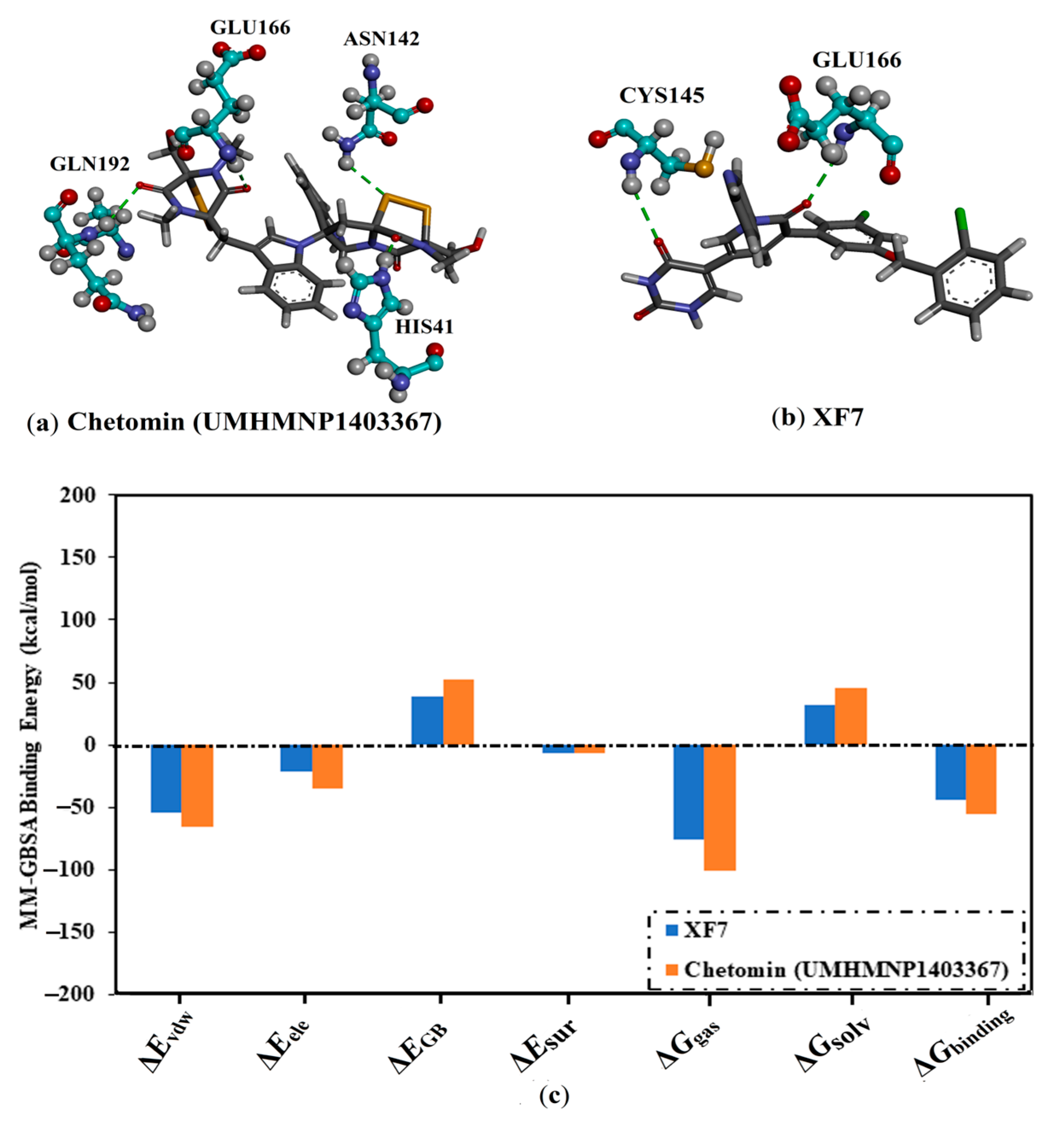
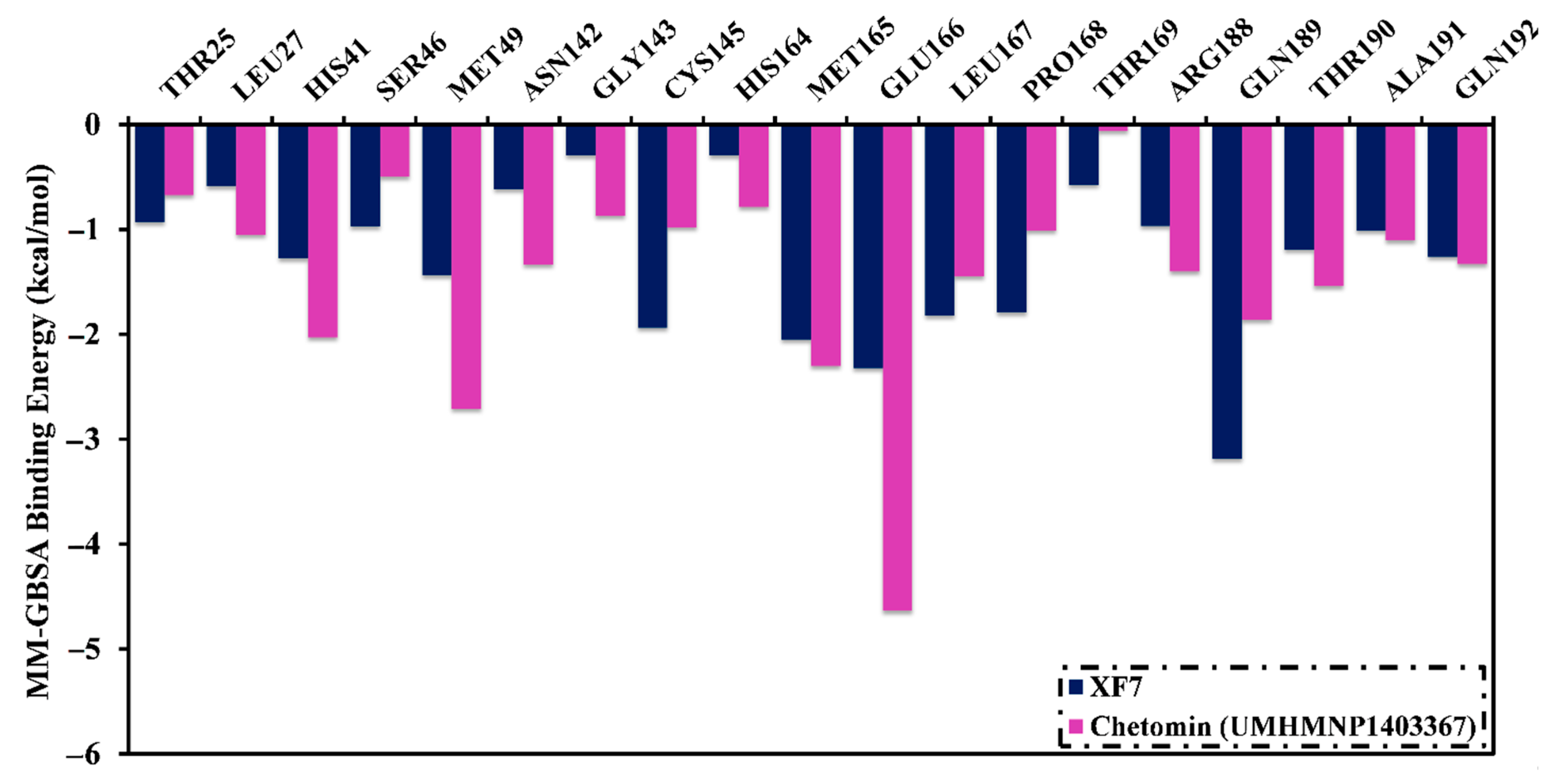
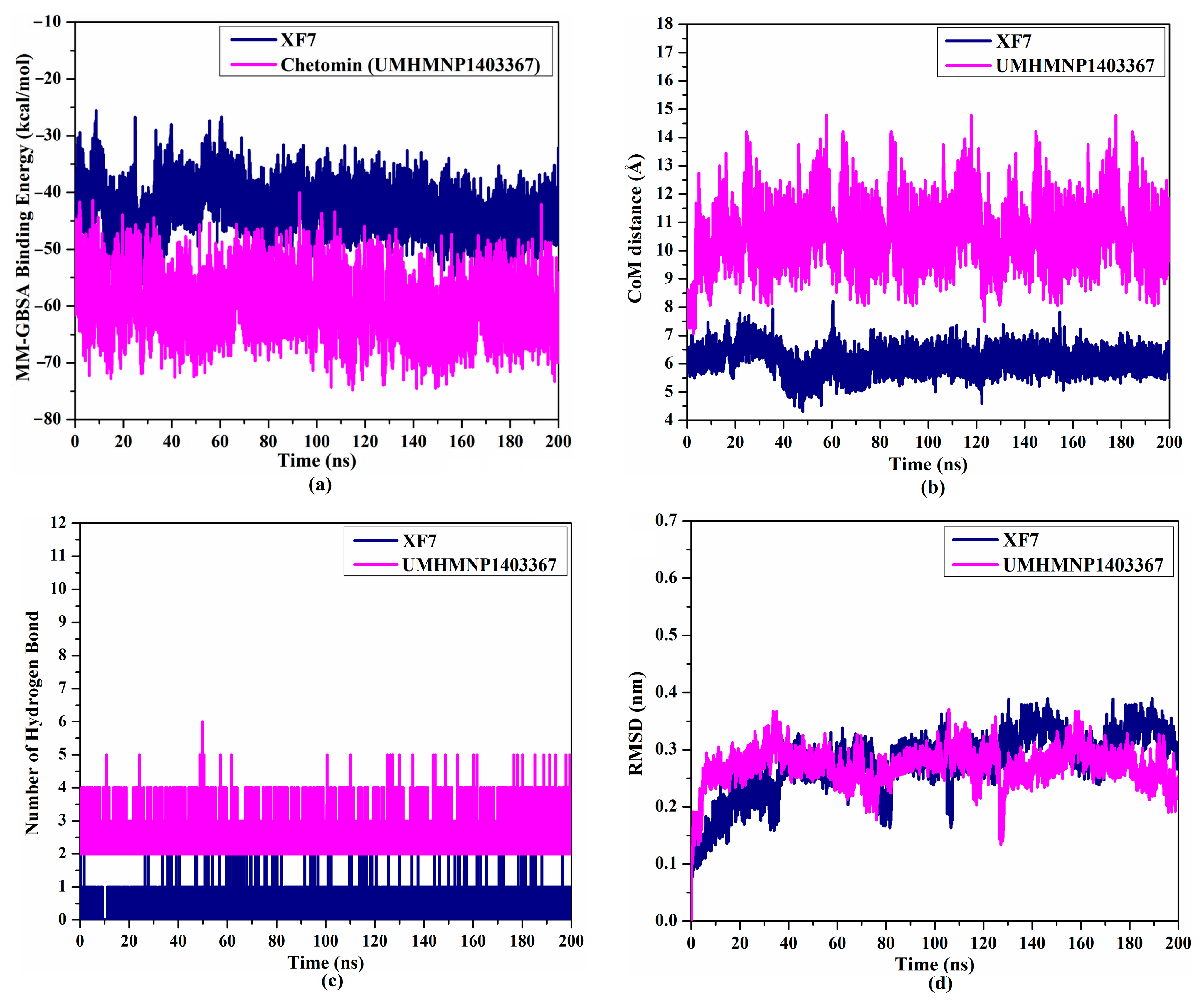
3.4.1. Binding Affinity Analysis
3.4.2. CoM Distance
3.4.3. H-Bond Numbers
3.4.4. Root-Mean-Square Deviation
3.5. Physicochemical Features
3.6. Pharmacokinetic Characteristics
3.7. Chetomin Derivatives as Prospective Anti-COVID-19 Drug Candidates
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef]
- Sohrabi, C.; Alsafi, Z.; O’Neill, N.; Khan, M.; Kerwan, A.; Al-Jabir, A.; Iosifidis, C.; Agha, R. World Health Organization declares global emergency: A review of the 2019 novel coronavirus (COVID-19). Int. J. Surg. 2020, 76, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [PubMed]
- She, J.; Jiang, J.; Ye, L.; Hu, L.; Bai, C.; Song, Y. 2019 novel coronavirus of pneumonia in Wuhan, China: Emerging attack and management strategies. Clin. Transl. Med. 2020, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Mishra, N.P.; Das, S.S.; Yadav, S.; Khan, W.; Afzal, M.; Alarifi, A.; Kenawy, E.R.; Ansari, M.T.; Hasnain, M.S.; Nayak, A.K. Global impacts of pre- and post-COVID-19 pandemic: Focus on socio-economic consequences. Sens. Int. 2020, 1, 100042. [Google Scholar] [CrossRef]
- Telenti, A.; Arvin, A.; Corey, L.; Corti, D.; Diamond, M.S.; Garcia-Sastre, A.; Garry, R.F.; Holmes, E.C.; Pang, P.S.; Virgin, H.W. After the pandemic: Perspectives on the future trajectory of COVID-19. Nature 2021, 596, 495–504. [Google Scholar] [CrossRef]
- Astuti, I.; Ysrafil. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): An overview of viral structure and host response. Diabetes Metab. Syndr. 2020, 14, 407–412. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef]
- Li, Q.; Kang, C. Progress in Developing Inhibitors of SARS-CoV-2 3C-Like Protease. Microorganisms 2020, 8, 1250. [Google Scholar] [CrossRef]
- Suarez, D.; Diaz, N. SARS-CoV-2 Main Protease: A Molecular Dynamics Study. J. Chem. Inf. Model. 2020, 60, 5815–5831. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.A.A.; Abdelrahman, A.H.M.; Hegazy, M.F. In-silico drug repurposing and molecular dynamics puzzled out potential SARS-CoV-2 main protease inhibitors. J. Biomol. Struct. Dyn. 2021, 39, 5756–5767. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.A.A.; Abdelrahman, A.H.M.; Allemailem, K.S.; Almatroudi, A.; Moustafa, M.F.; Hegazy, M.F. In Silico evaluation of prospective anti-COVID-19 drug candidates as potential SARS-CoV-2 main protease inhibitors. Protein J. 2021, 40, 296–309. [Google Scholar] [CrossRef] [PubMed]
- Pawar, A.Y. Combating devastating COVID-19 by drug repurposing. Int. J. Antimicrob. Agents 2020, 56, 105984. [Google Scholar] [CrossRef]
- Drozdzal, S.; Rosik, J.; Lechowicz, K.; Machaj, F.; Kotfis, K.; Ghavami, S.; Los, M.J. FDA approved drugs with pharmacotherapeutic potential for SARS-CoV-2 (COVID-19) therapy. Drug Resist. Updates 2020, 53, 100719. [Google Scholar] [CrossRef]
- Musarra-Pizzo, M.; Pennisi, R.; Ben-Amor, I.; Mandalari, G.; Sciortino, M.T. Antiviral Activity Exerted by Natural Products against Human Viruses. Viruses 2021, 13, 828. [Google Scholar] [CrossRef]
- Lin, L.T.; Hsu, W.C.; Lin, C.C. Antiviral natural products and herbal medicines. J. Tradit. Complement. Med. 2014, 4, 24–35. [Google Scholar] [CrossRef]
- Lichtiger, S.; Present, D.H.; Kornbluth, A.; Gelernt, I.; Bauer, J.; Galler, G.; Michelassi, F.; Hanauer, S. Cyclosporine in severe ulcerative colitis refractory to steroid therapy. N. Engl. J. Med. 1994, 330, 1841–1845. [Google Scholar] [CrossRef]
- Magnusson, I.; Ekman, L.; Wangdahl, M.; Wahren, J. N-Acetyl-L-Tyrosine and N-Acetyl-L-Cysteine as Tyrosine and Cysteine Precursors during Intravenous-Infusion in Humans. Metab. Clin. Exp. 1989, 38, 957–961. [Google Scholar] [CrossRef]
- Mia, M.M.; Hasan, M.; Miah, M.M.; Hossain, M.A.S.; Islam, S.; Shanta, V. Inhibitory Potentiality of Secondary Metabolites Extracted from Marine Fungus Target on Avian Influenza Virus-A Subtype H5N8 (Neuraminidase) and H5N1 (Nucleoprotein): A Rational Virtual Screening. Vet. Anim. Sci. 2022, 15, 100231. [Google Scholar] [CrossRef]
- Banerjee, P.; Mandhare, A.; Bagalkote, V. Marine natural products as source of new drugs: An updated patent review (July 2018–July 2021). Expert Opin. Ther. Pat. 2022, 32, 317–363. [Google Scholar] [CrossRef]
- Choudhary, A.; Naughton, L.M.; Montanchez, I.; Dobson, A.D.W.; Rai, D.K. Current Status and Future Prospects of Marine Natural Products (MNPs) as Antimicrobials. Mar. Drugs 2017, 15, 272. [Google Scholar] [CrossRef] [PubMed]
- Riccio, G.; Ruocco, N.; Mutalipassi, M.; Costantini, M.; Zupo, V.; Coppola, D.; de Pascale, D.; Lauritano, C. Ten-Year Research Update Review: Antiviral Activities from Marine Organisms. Biomolecules 2020, 10, 1007. [Google Scholar] [CrossRef] [PubMed]
- Yasuhara-Bell, J.; Lu, Y. Marine compounds and their antiviral activities. Antivir. Res. 2010, 86, 231–240. [Google Scholar] [CrossRef]
- Gogineni, V.; Schinazi, R.F.; Hamann, M.T. Role of Marine Natural Products in the Genesis of Antiviral Agents. Chem. Rev. 2015, 115, 9655–9706. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Kim, J.H.; Kwon, J.M.; Kwon, H.J.; Jeong, H.J.; Kim, Y.M.; Kim, D.; Lee, W.S.; Ryu, Y.B. Dieckol, a SARS-CoV 3CL(pro) inhibitor, isolated from the edible brown algae Ecklonia cava. Bioorganic Med. Chem. 2013, 21, 3730–3737. [Google Scholar] [CrossRef]
- Lira, S.P.d.; Seleghim, M.H.R.; Williams, D.E.; Marion, F.; Hamill, P.; Jean, F.; Andersen, R.J.; Hajdu, E.; Berlinck, R.G.S. A SARS-coronovirus 3CL protease inhibitor isolated from the marine sponge Axinella cf. corrugata: Structure elucidation and synthesis. J. Braz. Chem. Soc. 2007, 18, 440–443. [Google Scholar] [CrossRef]
- Zhang, C.H.; Stone, E.A.; Deshmukh, M.; Ippolito, J.A.; Ghahremanpour, M.M.; Tirado-Rives, J.; Spasov, K.A.; Zhang, S.; Takeo, Y.; Kudalkar, S.N.; et al. Potent Noncovalent Inhibitors of the Main Protease of SARS-CoV-2 from Molecular Sculpting of the Drug Perampanel Guided by Free Energy Perturbation Calculations. ACS Cent. Sci. 2021, 7, 467–475. [Google Scholar] [CrossRef]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005, 33, W368–W371. [Google Scholar] [CrossRef]
- OMEGA, Version 2.5.1.4; OpenEye Scientific Software: Santa Fe, NM, USA, 2013.
- Hawkins, P.C.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer generation with OMEGA: Algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef]
- QUACPAC, Version 1.7.0.2; OpenEye Scientific Software: Santa Fe, NM, USA, 2016.
- Gasteiger, J.; Marsili, M. Iterative Partial Equalization of Orbital Electronegativity—A Rapid Access to Atomic Charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Heller, S.R.; McNaught, A.; Pletnev, I.; Stein, S.; Tchekhovskoi, D. InChI, the IUPAC International Chemical Identifier. J. Cheminformatics 2015, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- McGann, M. FRED pose prediction and virtual screening accuracy. J. Chem. Inf. Model. 2011, 51, 578–596. [Google Scholar] [CrossRef]
- McGann, M. FRED and HYBRID docking performance on standardized datasets. J. Comput. Aided Mol. Des. 2012, 26, 897–906. [Google Scholar] [CrossRef]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Case, D.A.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E.; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer, N.; et al. AMBER 2016; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Ibrahim, M.A.A.; Abdeljawaad, K.A.A.; Abdelrahman, A.H.M.; Alzahrani, O.R.; Alshabrmi, F.M.; Khalaf, E.; Moustafa, M.F.; Alrumaihi, F.; Allemailem, K.S.; Soliman, M.E.S.; et al. Non-beta-lactam allosteric inhibitors target methicillin-resistant staphylococcus aureus: An in Silico drug discovery study. Antibiotics 2021, 10, 934. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Abdelrahman, A.H.M.; Badr, E.A.A.; Almansour, N.M.; Alzahrani, O.R.; Ahmed, M.N.; Soliman, M.E.S.; Naeem, M.A.; Shawky, A.M.; Sidhom, P.A.; et al. Naturally occurring plant-based anticancerous candidates as prospective ABCG2 inhibitors: An in silico drug discovery study. Mol. Divers. 2022, 26, 3255–3277. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Abdelrahman, A.H.M.; Hassan, A.M.A. Identification of novel Plasmodium falciparum PI4KB inhibitors as potential anti-malarial drugs: Homology modeling, molecular docking and molecular dynamics simulations. Comput. Biol. Chem. 2019, 80, 79–89. [Google Scholar] [CrossRef]
- Ibrahim, M.A.A.; Badr, E.A.A.; Abdelrahman, A.H.M.; Almansour, N.M.; Mekhemer, G.A.H.; Shawky, A.M.; Moustafa, M.F.; Atia, M.A.M. In Silico targeting human multidrug transporter ABCG2 in breast cancer: Database screening, molecular docking, and molecular dynamics study. Mol. Inform. 2022, 41, e2060039. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.A.A.; Badr, E.A.A.; Abdelrahman, A.H.M.; Almansour, N.M.; Shawky, A.M.; Mekhemer, G.A.H.; Alrumaihi, F.; Moustafa, M.F.; Atia, M.A.M. Prospective drug candidates as human multidrug transporter ABCG2 inhibitors: An in Silico drug discovery study. Cell Biochem. Biophys. 2021, 79, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef] [PubMed]
- Roux, B.; Simonson, T. Implicit solvent models. Biophys. Chem. 1999, 78, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision E01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Bayly, C.I.; Cieplak, P.; Cornell, W.D.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges-the RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: AnN⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle-An Analytical Version of the Shake and Rattle Algorithm for Rigid Water Models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Dassault Systèmes BIOVIA. Discovery Studio Visualize, Version 2019; Dassault Systèmes BIOVIA: San Diego, CA, USA, 2019. [Google Scholar]
- Massova, I.; Kollman, P.A. Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspect. Drug Discov. 2000, 18, 113–135. [Google Scholar] [CrossRef]
- Weiser, J.; Shenkin, P.S.; Still, W.C. Approximate atomic surfaces from linear combinations of pairwise overlaps (LCPO). J. Comput. Chem. 1999, 20, 217–230. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the molecular mechanics/Poisson Boltzmann surface area and molecular mechanics/generalized Born surface area methods. II. The accuracy of ranking poses generated from docking. J. Comput. Chem. 2011, 32, 866–877. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-point binding free energy calculation with MM/PBSA and MM/GBSA: Strategies and applications in drug design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Geahchan, S.; Ehrlich, H.; Rahman, M.A. The Anti-Viral Applications of Marine Resources for COVID-19 Treatment: An Overview. Mar. Drugs 2021, 19, 409. [Google Scholar] [CrossRef]
- De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of molecular dynamics and related methods in drug discovery. J. Med. Chem. 2016, 59, 4035–4061. [Google Scholar] [CrossRef]
- Kerrigan, J.E. Molecular dynamics simulations in drug design. In In Silico Models for Drug Discovery; Kortagere, S., Ed.; Humana Press: Totowa, NJ, USA, 2013; pp. 95–113. [Google Scholar]
- Mullard, A. Re-assessing the rule of 5, two decades on. Nat. Rev. Drug Discov. 2018, 17, 777. [Google Scholar] [CrossRef]
- Han, Y.; Zhang, J.; Hu, C.Q.; Zhang, X.; Ma, B.; Zhang, P. In silico ADME and toxicity prediction of ceftazidime and its impurities. Front. Pharmacol. 2019, 10, 434–443. [Google Scholar] [CrossRef]
- Dahlgren, D.; Lennernas, H. Intestinal Permeability and Drug Absorption: Predictive Experimental, Computational and In Vivo Approaches. Pharmaceutics 2019, 11, 411. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Compound ID | Docking Score (kcal/mol) | 2D Chemical Structure | Binding Feature b | ||
|---|---|---|---|---|---|---|
| Fast | Moderate | Expensive | ||||
| XF7 | −8.1 | −9.2 | −9.5 |  | THR26 (2.28, 2.49 Å), GLY143 (2.07 Å), SER144 (3.29 Å), HIS163 (1.91 Å), GLU166 (1.81 Å) | |
| 1 | UMHMNP1403367 (Chemotin) | −11.7 | −12.2 | −13.4 |  | GLU166 (1.99, 2.79 Å) |
| 2 | UMHMNP101691127 | −11.4 | −12.1 | −12.3 |  | LEU141 (2.01, 2.22 Å), CYS145 (2.33 Å), GLU166 (2.01, 2.84 Å), GLN189 (1.89 Å) |
| 3 | UMHMNP791849666 | −11.4 | −11.9 | −12.3 |  | ASN142 (1.96 Å), HIS163 (1.93 Å), GLU166 (2.01, 2.09 Å), ARG188 (1.78 Å) |
| 4 | UMHMNP14984668 | −11.3 | −11.7 | −12.2 |  | HIS41 (2.41 Å), GLY143 (2.31 Å), SER144 (2.28 Å), CYS145 (2.09, 2.17 Å), ARG188 (2.00 Å) |
| 5 | UMHMNP143621754 | −11.3 | −11.7 | −12.2 |  | HIS41 (2.14 Å), GLY143 (2.75, 2.91 Å), CYS145 (2.45 Å), GLU166 (2.10, 2.78 Å) |
| 6 | UMHMNP148839036 | −11.3 | −11.6 | −11.8 |  | HIS41 (2.20 Å), LEU141 (2.09 Å), CYS145 (2.14, 2.27 Å) |
| 7 | UMHMNP133056072 | −11.3 | −11.6 | −11.7 |  | GLU166 (2.30 Å) |
| 8 | UMHMNP386274857 | −11.3 | −11.6 | −11.7 |  | SER144 (1.91 Å), CYS145 (2.41 Å), GLN192 (2.13 Å) |
| 9 | UMHMNP133056094 | −11.2 | −11.5 | −11.5 |  | GLU166 (2.82 Å), THR190 (2.56 Å), GLN192 (2.48 Å) |
| 10 | UMHMNP26195584 | −11.1 | −11.5 | −11.5 |  | HIS28 (2.28 Å), GLY143 (2.51 Å), CYS145 (2.15, 2.72 Å), HIS163 (1.92 Å), GLN189 (2.29 Å) |
| 11 | UMHMNP874383707 | −11.1 | −11.5 | −11.4 |  | LEU141 (2.08 Å), GLY143 (2.75 Å), SER144 (2.09 Å), CYS145 (2.46 Å), GLU166 (1.75 Å) |
| 12 | UMHMNP221163300 | −11.1 | −11.4 | −11.4 |  | PHE140 (2.04 Å), GLY143 (2.32 Å), CYS145 (2.87 Å), HIS163 (1.92 Å) |
| 13 | UMHMNP109152387 | −11.1 | −11.4 | −11.4 |  | HIS41 (2.25 Å), PHE140 (1.93 Å), GLY143 (2.44 Å), CYS145 (2.35, 2.36 Å), HIS163 (2.00 Å), GLU166 (2.57 Å), HIS172 (2.23 Å) |
| Compound ID/Name | Log po/w | MWt | HBD | HBA | TPSA | Nrotb |
|---|---|---|---|---|---|---|
| XF7 | 4.25 | 533.4 | 2 | 5 | 109.8 | 6 |
| Chetomin | 1.05 | 710.9 | 3 | 6 | 239.9 | 5 |
| ADMET Characteristics | Chetomin (UMHMNP1403367) | XF7 |
|---|---|---|
| Absorption (A) | ||
| Skin permeability (log Kp) | −2.7 | −2.7 |
| Caco2 permeability (log Papp, cm/s) | −0.05 | 0.5 |
| Intestinal absorption (human) (%) | 73.4% | 93.5% |
| P-glycoprotein substrate (Yes/No) | Yes | Yes |
| P-glycoprotein I inhibitor (Yes/No) | Yes | Yes |
| P-glycoprotein II inhibitor (Yes/No) | Yes | Yes |
| Distribution (D) | ||
| VDss (human) (log L/kg) | −0.1 | 0.3 |
| BBB permeability (log BB) | −2.0 | −1.1 |
| CNS permeability (log PS) | −3.9 | −2.5 |
| Metabolism (M) | ||
| CYP1A2 inhibitor (Yes/No) | No | No |
| CYP2C19 inhibitor (Yes/No) | No | Yes |
| CYP2C9 inhibitor (Yes/No) | No | Yes |
| CYP2D6 inhibitor (Yes/No) | No | No |
| CYP3A4 inhibitor (Yes/No) | No | Yes |
| Excretion (E) | ||
| Total Clearance (log mL/min/kg) | 0.3 | 0.8 |
| Toxicity (T) | ||
| AMES toxicity (Yes/No) | No | No |
| Skin Sensitization (Yes/No) | No | No |
| No. | Compound Name/PubChem ID | Docking Score (kcal/mol) | 2D Chemical Structures | Binding Features a |
|---|---|---|---|---|
| 1 | PubChem10417379 (Chetomin) | −13.4 |  | GLU166 (1.99, 2.79 Å) |
| 1 | PubChem139591137 (Chetomin B) | −12.2 |  | GLU166 (2.69 Å), CYS145 (2.12 Å), HIS41 (1.71 Å) |
| 2 | PubChem139591139 (Chetomin D) | −12.0 |  | PRO168 (2.07 Å), GLU166 (1.83 Å), CYS145 (2.56, 2.95 Å), LEU141 (1.97 Å) |
| 3 | PubChem139591136 (Chetomin A) | −11.5 |  | ASN142 (1.88, 2.93, 3.04 Å) |
| 4 | PubChem139591138 (Chetomin C) | −11.1 |  | GLN192 (2.46 Å), THR190 (1.88 Å), GLU166 (3.10 Å), ASN142 (2.17 Å) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibrahim, M.A.A.; Abdelrahman, A.H.M.; Mohamed, D.E.M.; Abdeljawaad, K.A.A.; Naeem, M.A.; Gabr, G.A.; Shawky, A.M.; Soliman, M.E.S.; Sidhom, P.A.; Paré, P.W.; et al. Chetomin, a SARS-CoV-2 3C-like Protease (3CLpro) Inhibitor: In Silico Screening, Enzyme Docking, Molecular Dynamics and Pharmacokinetics Analysis. Viruses 2023, 15, 250. https://doi.org/10.3390/v15010250
Ibrahim MAA, Abdelrahman AHM, Mohamed DEM, Abdeljawaad KAA, Naeem MA, Gabr GA, Shawky AM, Soliman MES, Sidhom PA, Paré PW, et al. Chetomin, a SARS-CoV-2 3C-like Protease (3CLpro) Inhibitor: In Silico Screening, Enzyme Docking, Molecular Dynamics and Pharmacokinetics Analysis. Viruses. 2023; 15(1):250. https://doi.org/10.3390/v15010250
Chicago/Turabian StyleIbrahim, Mahmoud A. A., Alaa H. M. Abdelrahman, Dina E. M. Mohamed, Khlood A. A. Abdeljawaad, Mohamed Ahmed Naeem, Gamal A. Gabr, Ahmed M. Shawky, Mahmoud E. S. Soliman, Peter A. Sidhom, Paul W. Paré, and et al. 2023. "Chetomin, a SARS-CoV-2 3C-like Protease (3CLpro) Inhibitor: In Silico Screening, Enzyme Docking, Molecular Dynamics and Pharmacokinetics Analysis" Viruses 15, no. 1: 250. https://doi.org/10.3390/v15010250
APA StyleIbrahim, M. A. A., Abdelrahman, A. H. M., Mohamed, D. E. M., Abdeljawaad, K. A. A., Naeem, M. A., Gabr, G. A., Shawky, A. M., Soliman, M. E. S., Sidhom, P. A., Paré, P. W., & Hegazy, M.-E. F. (2023). Chetomin, a SARS-CoV-2 3C-like Protease (3CLpro) Inhibitor: In Silico Screening, Enzyme Docking, Molecular Dynamics and Pharmacokinetics Analysis. Viruses, 15(1), 250. https://doi.org/10.3390/v15010250