
Hazara Orthonairovirus Nucleoprotein Antagonizes Type I Interferon Production by Inhibition of RIG-I Ubiquitination
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Virus
2.2. Antibodies and Reagents
2.3. Plasmids
2.4. Plaque Assay
2.5. Immunoblot and Immunoprecipitation Assays
2.6. RIG-I Ubiquitination Assay
2.7. Type I IFN Detection (Secreted Embryonic Alkaline Phosphatase (SEAP) Assay)
3. Results
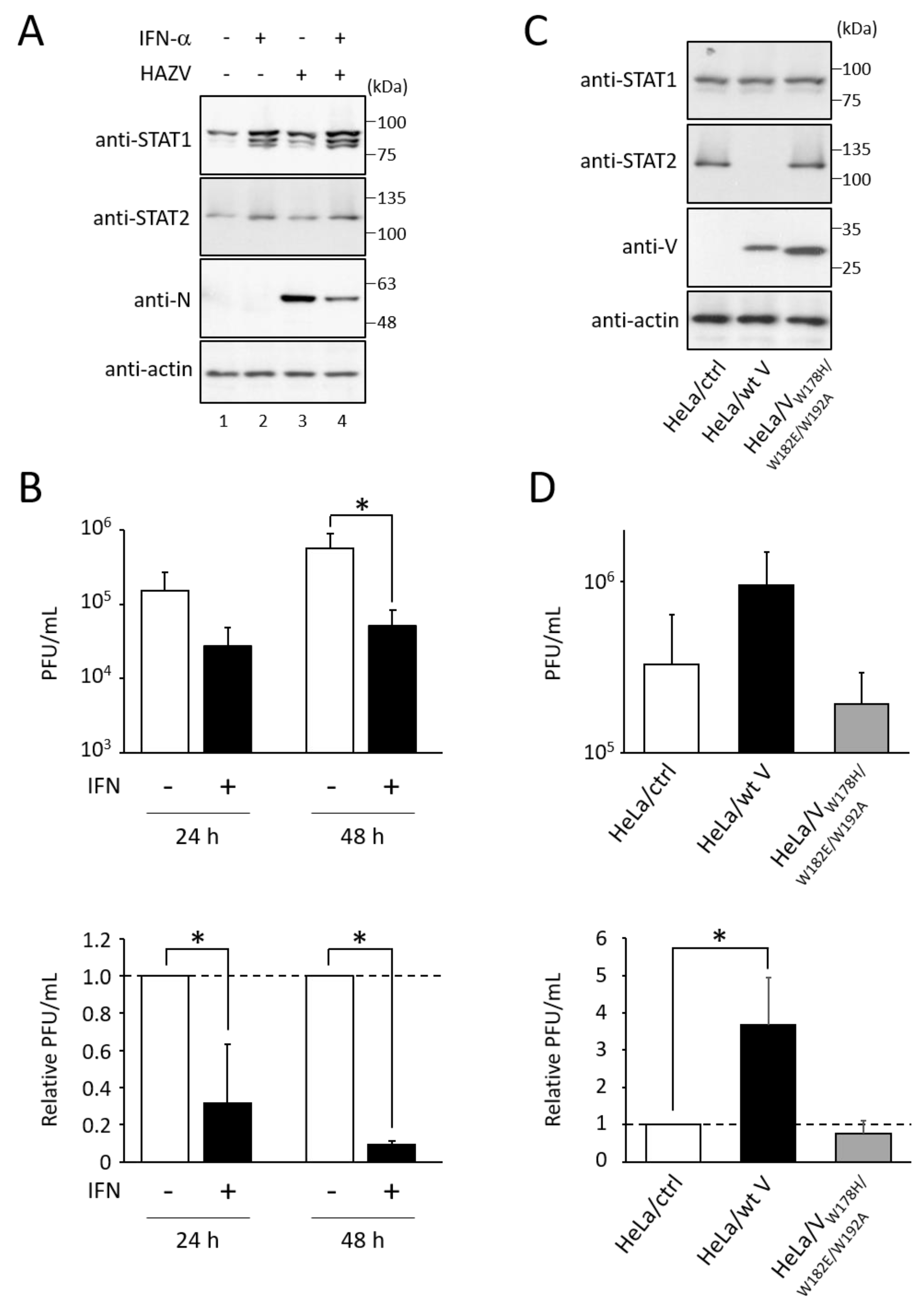
3.1. Type I IFN Inhibits the Growth of HAZV
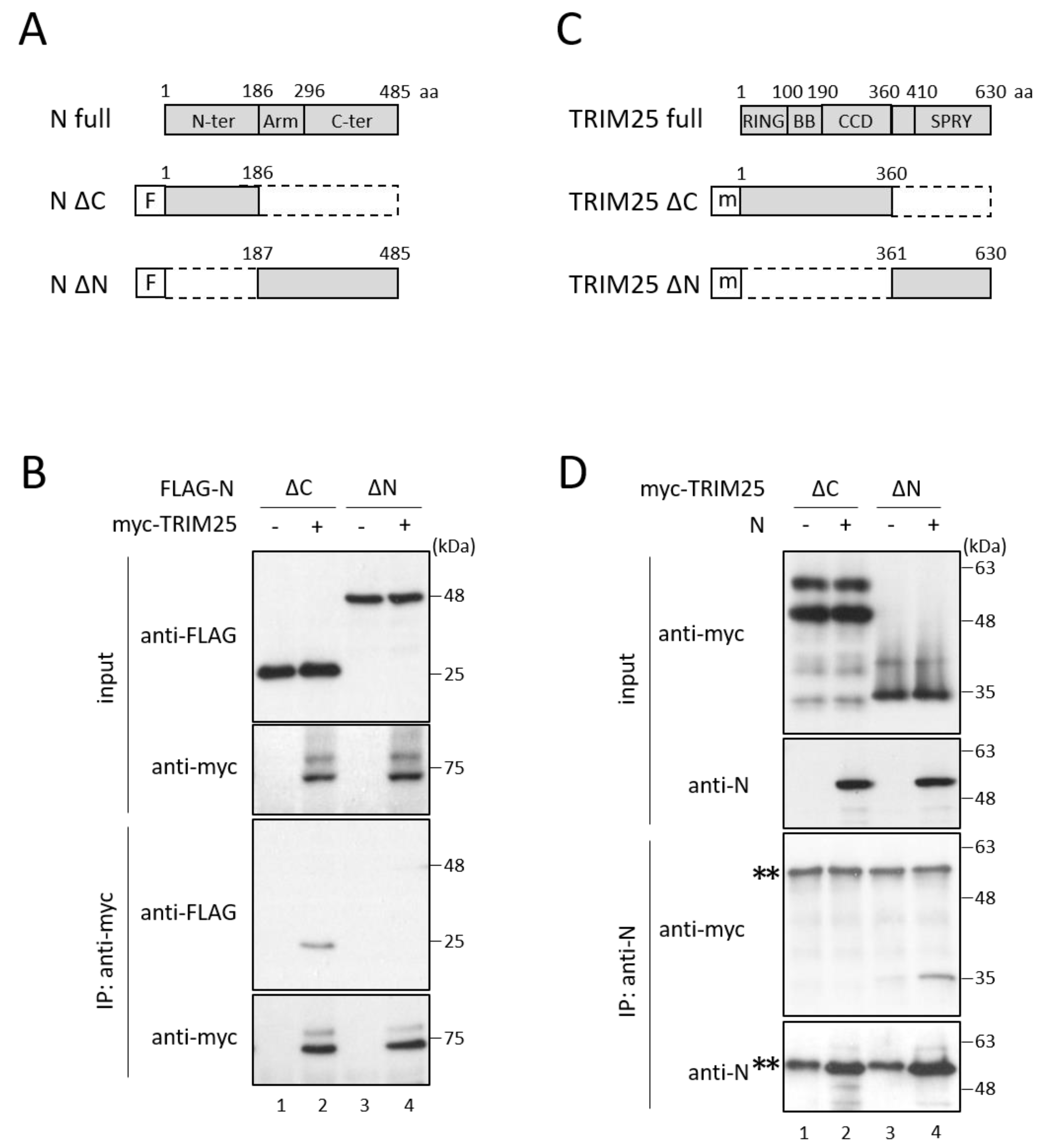
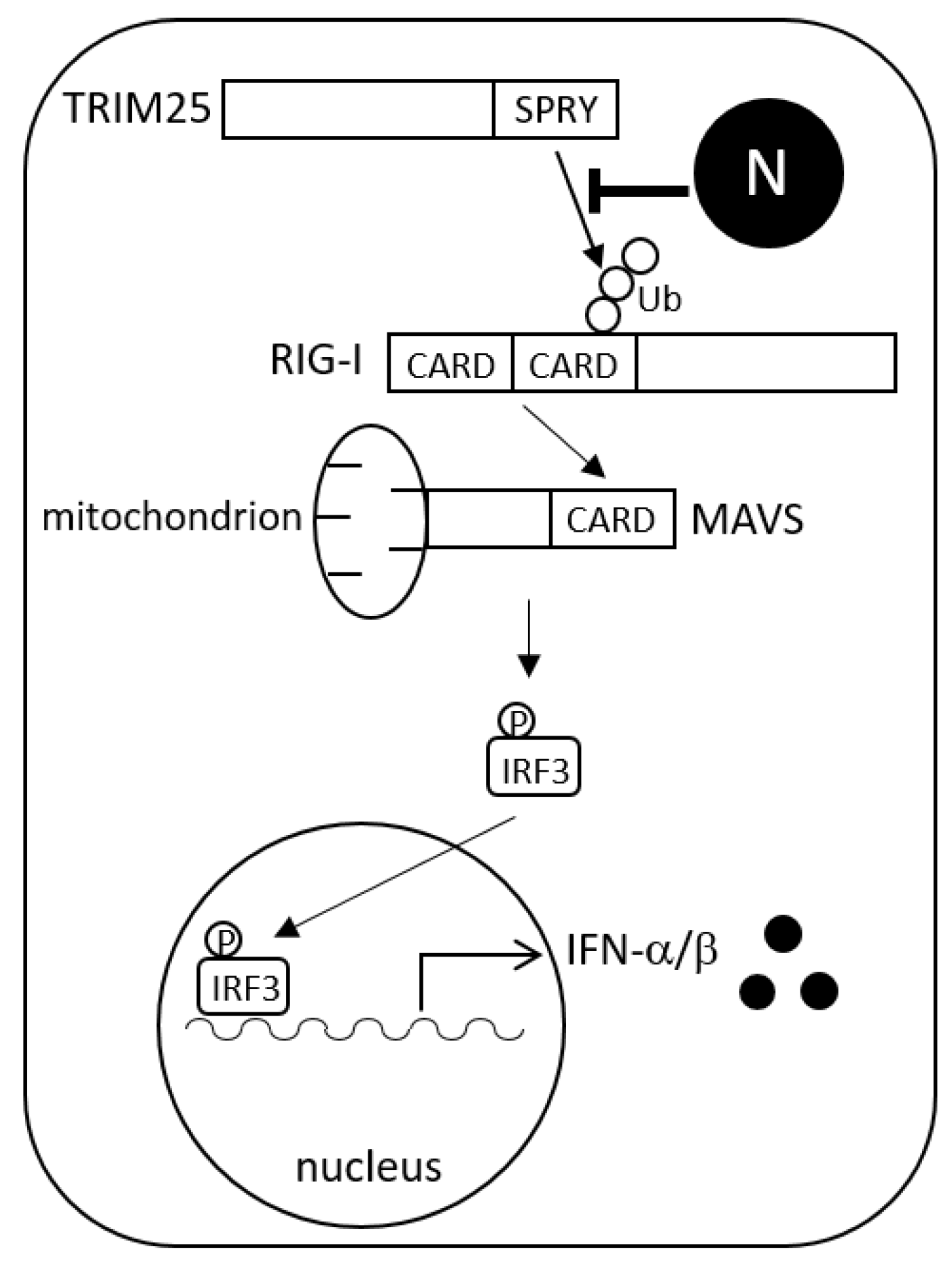
3.2. HAZV N Protein Binds to TRIM25
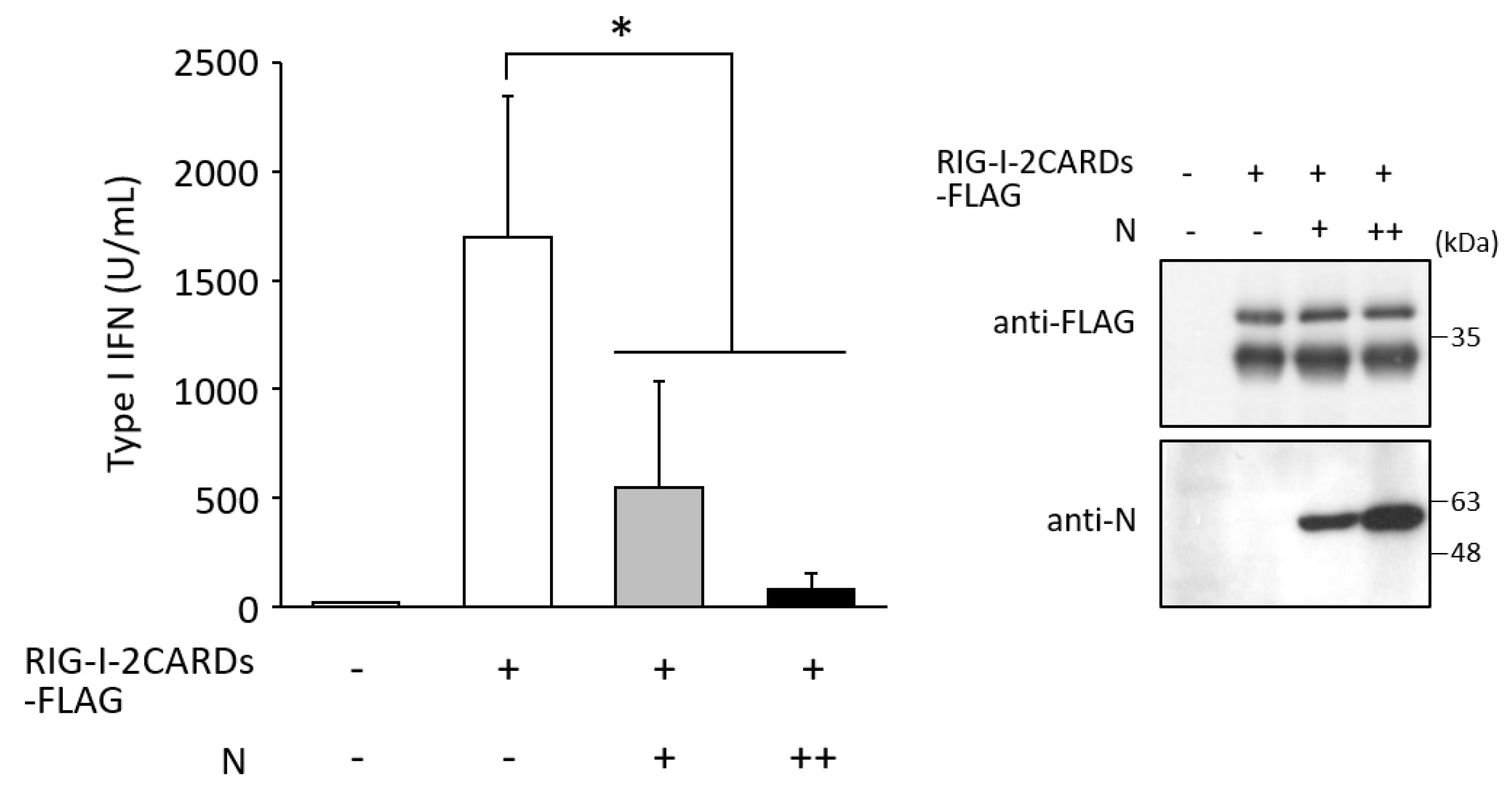
3.3. N Protein Inhibits RIG-I Ubiquitination
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Elliott, R.M.; Schmaljohn, C. Bunyaviridae. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Cohen, J.I., Griffin, D.E., Lamb, R.A., Martin, M.A., Racaniello, V.R., Roizman, B., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1244–1282. [Google Scholar]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef]
- Yoneyama, M.; Fujita, T. RIG-I family RNA helicases: Cytoplasmic sensor for antiviral innate immunity. Cytokine Growth Factor Rev. 2007, 18, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Uematsu, S.; Matsui, K.; Tsujimura, T.; Takeda, K.; Fujita, T.; Takeuchi, O.; et al. Cell type-specific involvement of RIG-I in antiviral response. Immunity 2005, 23, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Errett, J.S.; Gale, M. Emerging complexity and new roles for the RIG-I-like receptors in innate antiviral immunity. Virol. Sin. 2015, 30, 163–173. [Google Scholar] [CrossRef]
- Meylan, E.; Tschopp, J.; Karin, M. Intracellular pattern recognition receptors in the host response. Nature 2006, 442, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Gack, M.U.; Shin, Y.C.; Joo, C.H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007, 446, 916–920. [Google Scholar] [CrossRef]
- Oshiumi, H.; Matsumoto, M.; Hatakeyama, S.; Seya, T. Riplet/RNF135, a RING finger protein, ubiquitinates RIG-I to promote interferon-beta induction during the early phase of viral infection. J. Biol. Chem. 2009, 284, 807–817. [Google Scholar] [CrossRef]
- Lang, X.; Tang, T.; Jin, T.; Ding, C.; Zhou, R.; Jiang, W. TRIM65-catalized ubiquitination is essential for MDA5-mediated antiviral innate immunity. J. Exp. Med. 2017, 214, 459–473. [Google Scholar] [CrossRef]
- Rehwinkel, J.; Reis e Sousa, C. RIGorous detection: Exposing virus through RNA sensing. Science 2010, 327, 284–286. [Google Scholar] [CrossRef]
- Andrejeva, J.; Childs, K.S.; Young, D.F.; Carlos, T.S.; Stock, N.; Goodbourn, S.; Randall, R.E. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc. Natl. Acad. Sci. USA 2004, 101, 17264–17269. [Google Scholar] [CrossRef] [Green Version]
- Childs, K.; Randall, R.; Goodbourn, S. Paramyxovirus V proteins interact with the RNA Helicase LGP2 to inhibit RIG-I-dependent interferon induction. J. Virol. 2012, 86, 3411–3421. [Google Scholar] [CrossRef] [PubMed]
- Ling, Z.; Tran, K.C.; Teng, M.N. Human respiratory syncytial virus nonstructural protein NS2 antagonizes the activation of beta interferon transcription by interacting with RIG-I. J. Virol. 2009, 83, 3734–3742. [Google Scholar] [CrossRef] [PubMed]
- Gack, M.U.; Albrecht, R.A.; Urano, T.; Inn, K.S.; Huang, I.C.; Carnero, E.; Farzan, M.; Inoue, S.; Jung, J.U.; García-Sastre, A. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 2009, 5, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Bouloy, M.; Janzen, C.; Vialat, P.; Khun, H.; Pavlovic, J.; Huerre, M.; Haller, O. Genetic evidence for an interferon-antagonistic function of Rift Valley fever virus nonstructural protein NSs. J. Virol. 2001, 75, 1371–1377. [Google Scholar] [CrossRef] [PubMed]
- Wuerth, J.D.; Weber, F. Phleboviruses and the Type I Interferon Response. Viruses 2016, 8, 174. [Google Scholar] [CrossRef]
- Qu, B.; Qi, X.; Wu, X.; Liang, M.; Li, C.; Cardona, C.J.; Xu, W.; Tang, F.; Li, Z.; Wu, B.; et al. Suppression of the interferon and NF-kappaB responses by severe fever with thrombocytopenia syndrome virus. J. Virol. 2012, 86, 8388–8401. [Google Scholar] [CrossRef] [PubMed]
- Min, Y.Q.; Ning, Y.J.; Wang, H.; Deng, F. A RIG-I-like receptor directs antiviral responses to a bunyavirus and is antagonized by virus-induced blockade of TRIM25-mediated ubiquitination. J. Biol. Chem. 2020, 295, 9691–9711. [Google Scholar] [CrossRef]
- Fan, L.; Briese, T.; Lipkin, W.I. Z proteins of New World arenaviruses bind RIG-I and interfere with type I interferon induction. J. Virol. 2010, 84, 1785–1791. [Google Scholar] [CrossRef]
- Bakshi, S.; Holzer, B.; Bridgen, A.; McMullan, G.; Quinn, D.G.; Baron, M.D. Dugbe virus ovarian tumour domain interferes with ubiquitin/ISG15-regulated innate immune cell signalling. J. Gen. Virol. 2013, 94, 298–307. [Google Scholar] [CrossRef]
- Scholte, F.E.M.; Zivcec, M.; Dzimianski, J.V.; Deaton, M.K.; Spengler, J.R.; Welch, S.R.; Nichol, S.T.; Pegan, S.D.; Spiropoulou, C.F.; Bergeron, É. Crimean-Congo Hemorrhagic Fever Virus Suppresses Innate Immune Responses via a Ubiquitin and ISG15 Specific Protease. Cell Rep. 2017, 20, 2396–2407. [Google Scholar] [CrossRef] [Green Version]
- Nishio, M.; Tsurudome, M.; Ito, M.; Garcin, D.; Kolakofsky, D.; Ito, Y. Identification of paramyxovirus V protein residues essential for STAT protein degradation and promotion of virus replication. J. Virol. 2005, 79, 8591–8601. [Google Scholar] [CrossRef] [PubMed]
- Begum, F.; Wisseman, C.L., Jr.; Casals, J. Tick-borne viruses of West Pakistan. II. Hazara virus, a new agent isolated from Ixodes redikorzevi ticks from the Kaghan Valley, W. Pakistan. Am. J. Epidemiol. 1970, 92, 192–194. [Google Scholar] [CrossRef] [PubMed]
- Nishio, M.; Tsurudome, M.; Ito, M.; Watanabe, N.; Kawano, M.; Komada, H.; Ito, Y. Human parainfluenza virus type 2 phosphoprotein: Mapping of monoclonal antibody epitopes and location of the multimerization domain. J. Gen. Virol. 1997, 78, 1303–1308. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y.; Nouchi, T.; Ohta, K.; Nishio, M. Regulation of Hazara virus growth through apoptosis inhibition by viral nucleoprotein. Arch. Virol. 2019, 164, 1597–1607. [Google Scholar] [CrossRef] [PubMed]
- Komada, H.; Kusagawa, S.; Orvell, C.; Tsurudome, M.; Nishio, M.; Bando, H.; Kawano, M.; Matsumura, H.; Norrby, E.; Ito, Y. Antigenic diversity of human parainfluenza virus type 1 isolates and their immunological relationship with Sendai virus revealed by using monoclonal antibodies. J. Gen. Virol. 1992, 73, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Surtees, R.; Ariza, A.; Punch, E.K.; Trinh, C.H.; Dowall, S.D.; Hewson, R.; Hiscox, J.A.; Barr, J.N.; Edwards, T.A. The crystal structure of the Hazara virus nucleocapsid protein. BMC Struct. Biol. 2015, 15, 24. [Google Scholar] [CrossRef]
- Sanchez, J.G.; Chiang, J.J.; Sparrer, K.M.J.; Alam, S.L.; Chi, M.; Roganowicz, M.D.; Sankaran, B.; Gack, M.U.; Pornillos, O. Mechanism of TRIM25 Catalytic Activation in the Antiviral RIG-I Pathway. Cell Rep. 2016, 16, 1315–1325. [Google Scholar] [CrossRef]
- Cadena, C.; Ahmad, S.; Xavier, A.; Willemsen, J.; Park, S.; Park, J.W.; Oh, S.W.; Fujita, T.; Hou, F.; Binder, M.; et al. Ubiquitin-Dependent and -Independent Roles of E3 Ligase RIPLET in Innate Immunity. Cell 2019, 177, 1187–1200.e16. [Google Scholar] [CrossRef]
- Hayman, T.J.; Hsu, A.C.; Kolesnik, T.B.; Dagley, L.F.; Willemsen, J.; Tate, M.D.; Baker, P.J.; Kershaw, N.J.; Kedzierski, L.; Webb, A.I.; et al. RIPLET, and not TRIM25, is required for endogenous RIG-I-dependent antiviral responses. Immunol. Cell Biol. 2019, 97, 840–852. [Google Scholar] [CrossRef]
- Kato, K.; Ahmad, S.; Zhu, Z.; Young, J.M.; Mu, X.; Park, S.; Malik, H.S.; Hur, S. Structural analysis of RIG-I-like receptors reveals ancient rules of engagement between diverse RNA helicases and TRIM ubiquitin ligases. Mol. Cell 2021, 81, 599–613.e8. [Google Scholar] [CrossRef]
- Oshiumi, H.; Miyashita, M.; Matsumoto, M.; Seya, T. A distinct role of Riplet-mediated K63-Linked polyubiquitination of the RIG-I repressor domain in human antiviral innate immune responses. PLoS Pathog. 2013, 9, e1003533. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Li, W.; Gao, T.; Cui, Y.; Jin, Y.; Li, P.; Ma, Q.; Liu, X.; Cao, C. The severe acute respiratory syndrome coronavirus nucleocapsid inhibits type I interferon production by interfering with TRIM25-mediated RIG-I ubiquitination. J. Virol. 2017, 91, e02143-16. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.Y.; Liu, H.M.; Chang, M.F.; Chang, S.C. Middle East respiratory syndrome coronavirus nucleocapsid protein suppresses type I and type III interferon induction by targeting RIG-I signaling. J. Virol. 2020, 94, e00099-20. [Google Scholar] [CrossRef]
- Gori-Savellini, G.; Anichini, G.; Gandolfo, C.; Cusi, M.G. SARS-CoV-2 N Protein Targets TRIM25-Mediated RIG-I Activation to Suppress Innate Immunity. Viruses 2021, 13, 1439. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhu, Z.; Wang, C.; Yang, F.; Cao, W.; Li, P.; Du, X.; Zhao, F.; Liu, X.; Zheng, H. Foot-and-mouth disease virus 3B protein interacts with pattern recognition receptor RIG-I to block RIG-I-mediated immune signaling and inhibit host antiviral response. J. Immunol. 2020, 205, 2207–2221. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.; Pauli, E.K.; Biryukov, J.; Feister, K.F.; Meng, M.; White, E.A.; Münger, K.; Howley, P.M.; Meyers, C.; Gack, M.U. The human papillomavirus E6 oncoprotein targets USP15 and TRIM25 to suppress RIG-I-mediated innate immune signaling. J. Virol. 2018, 92, e01737-17. [Google Scholar] [CrossRef]
- Chiang, C.; Dvorkin, S.; Chiang, J.J.; Potter, R.B.; Gack, M.U. The small t antigen of JC virus antagonizes RIG-I-mediated innate immunity by inhibiting TRIM25’s RNA binding ability. mBio 2021, 12, e00620–e00621. [Google Scholar] [CrossRef]
- Dzimianski, J.V.; Mace, S.L.; Williams, I.L.; Freitas, B.T.; Pegan, S.D. Flipping the substrate preference of Hazara virus ovarian tumour domain protease through structure-based mutagenesis. Acta Crystallogr. D Struct. Biol. 2020, 76, 1114–1123. [Google Scholar] [CrossRef]
- Devignot, S.; Kromer, T.; Mirazimi, A.; Weber, F. ISG15 overexpression compensates the defect of Crimean-Congo hemorrhagic fever virus polymerase bearing a protease-inactive ovarian tumor domain. PLoS Negl. Trop. Dis. 2020, 14, e0008610. [Google Scholar] [CrossRef]
- Habjan, M.; Andersson, I.; Klingström, J.; Schümann, M.; Martin, A.; Zimmermann, P.; Wagner, V.; Pichlmair, A.; Schneider, U.; Mühlberger, E.; et al. Processing of genome 5’ termini as a strategy of negative-strand RNA viruses to avoid RIG-I-dependent interferon induction. PLoS ONE 2008, 3, e2032. [Google Scholar] [CrossRef]
- Spengler, J.R.; Patel, J.R.; Chakrabarti, A.K.; Zivcec, M.; García-Sastre, A.; Spiropoulou, C.F.; Bergeron, É. RIG-I Mediates an Antiviral Response to Crimean-Congo Hemorrhagic Fever Virus. J. Virol. 2015, 89, 10219–10229. [Google Scholar] [CrossRef] [PubMed]
- Blakqori, G.; Delhaye, S.; Habjan, M.; Blair, C.D.; Sánchez-Vargas, I.; Olson, K.E.; Attarzadeh-Yazdi, G.; Fragkoudis, R.; Kohl, A.; Kalinke, U.; et al. La Crosse bunyavirus nonstructural protein NSs serves to suppress the type I interferon system of mammalian hosts. J. Virol. 2007, 81, 4991–4999. [Google Scholar] [CrossRef] [PubMed]
- Barnwal, B.; Karlberg, H.; Mirazimi, A.; Tan, Y.J. The non-structural protein of Crimean-Congo hemorrhagic fever virus disrupts the mitochondrial membrane potential and induces apoptosis. J. Biol. Chem. 2016, 291, 582–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ohta, K.; Saka, N.; Nishio, M. Hazara Orthonairovirus Nucleoprotein Antagonizes Type I Interferon Production by Inhibition of RIG-I Ubiquitination. Viruses 2022, 14, 1965. https://doi.org/10.3390/v14091965
Ohta K, Saka N, Nishio M. Hazara Orthonairovirus Nucleoprotein Antagonizes Type I Interferon Production by Inhibition of RIG-I Ubiquitination. Viruses. 2022; 14(9):1965. https://doi.org/10.3390/v14091965
Chicago/Turabian StyleOhta, Keisuke, Naoki Saka, and Machiko Nishio. 2022. "Hazara Orthonairovirus Nucleoprotein Antagonizes Type I Interferon Production by Inhibition of RIG-I Ubiquitination" Viruses 14, no. 9: 1965. https://doi.org/10.3390/v14091965
APA StyleOhta, K., Saka, N., & Nishio, M. (2022). Hazara Orthonairovirus Nucleoprotein Antagonizes Type I Interferon Production by Inhibition of RIG-I Ubiquitination. Viruses, 14(9), 1965. https://doi.org/10.3390/v14091965