
Linking Human Betaretrovirus with Autoimmunity and Liver Disease in Patients with Primary Biliary Cholangitis


Abstract
1. Introduction
1.1. Primary Biliary Cholangitis
1.2. PBC: Epidemiology and Pathophysiology
1.3. PBC: Genes vs. Environment
1.4. PBC: Role of Autoimmunity
- Humoral autoimmunity does not propagate disease because AMA are not necessary to acquire PBC. AMA-negative patients develop the same disease process as AMA-positive individuals, and some AMA-positive individuals do not develop PBC [44]. No other autoantibodies are necessary to generate PBC.
- With regard to cellular immunity, PBC patients appear to have an insufficient precursor frequency of PDC-E2 reactive T lymphocytes to generate disease. The autoreactive T cells are estimated to be approximately 1 × 10−5 in the liver and 1 × 10−7 in the blood [45]. In comparison, preliminary studies suggest the frequency of HBRV reactive lymphocytes is approximately 100-fold higher in PBC patients with HBRV infection [46]. The level of anti-HBRV cellular immune responses is comparable with observations from other infectious diseases, such as chronic hepatitis C virus infection [47,48].
- While immunosuppression is the mainstay of treatment for autoimmune hepatitis, this therapeutic approach is of limited utility for PBC patients [49,50]. Moreover, liver transplant recipients are more likely to develop earlier and more severe recurrent PBC disease with the use of more potent immunosuppression regimens [51].
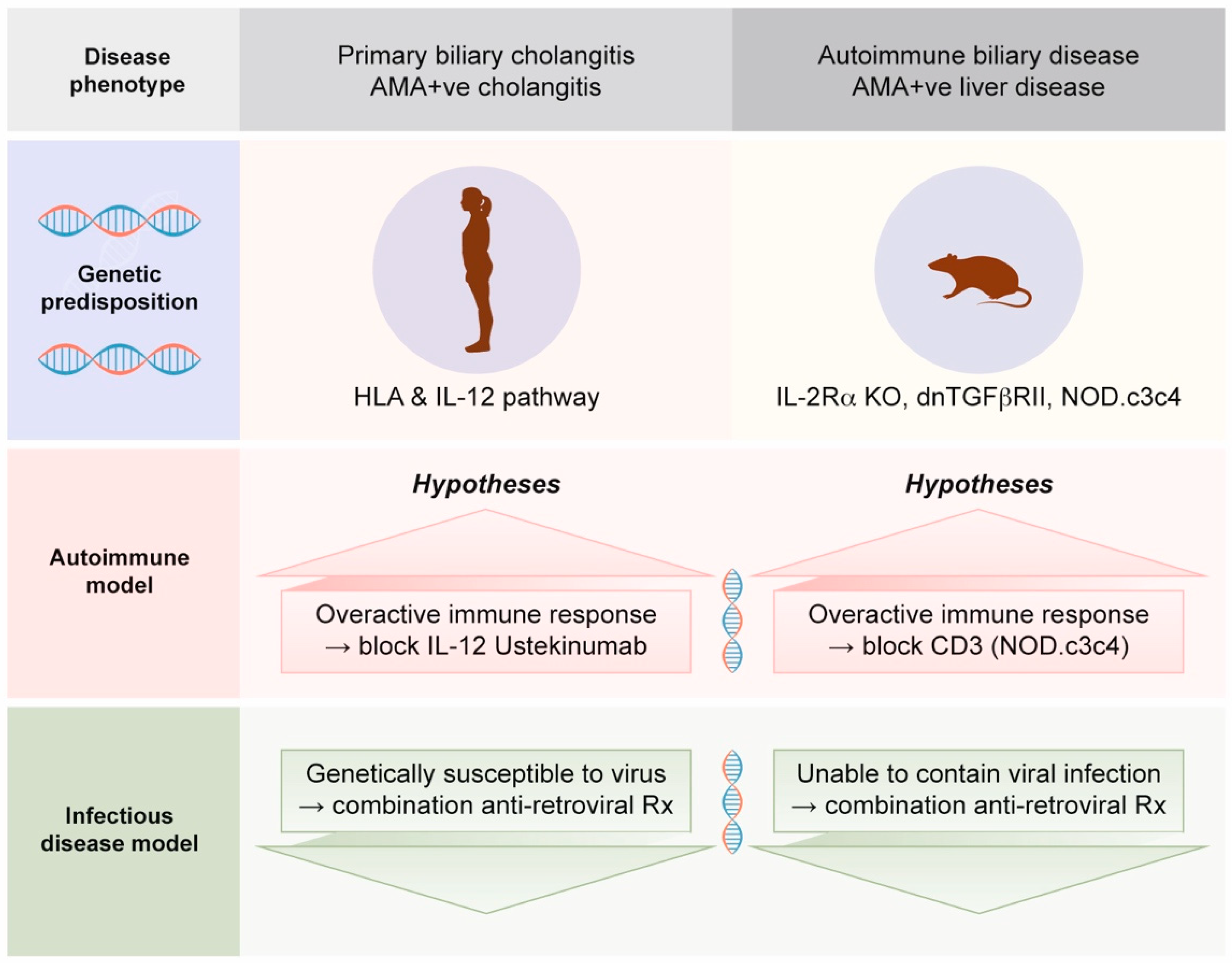
- Much of the assumption that PBC is an autoimmune disease is based on mouse models with autoimmune biliary disease that develop spontaneous AMA production [52]. Notably, these mouse models also have evidence of betaretrovirus infection with mouse mammary tumor virus (MMTV) protein expression [53]. We will address this process in Section 2.4, as betaretrovirus infection is directly linked with the production of AMA [7,54].
- In case series and clinical trials using combination ART to treat PBC patients, improvements in cholangitis have been linked with reduced viral load [56,57,58,59,60]. Notably, the gold standard of meeting randomized controlled trial study endpoints has not been achieved to date due to the lack of tolerability of combination ART and the lack of antiviral efficacy of repurposed ART used to treat HIV.
2. Human Betaretrovirus (HBRV)
2.1. Betaretroviruses
2.2. Mouse Mammary Tumor Virus (MMTV)
2.3. HBRV and Human Disorders
2.4. Challenges Linking HBRV with PBC
2.5. Linking HBRV with AMA Production
2.6. Linking MMTV with AMA Production in Mice
2.7. Linking MMTV with Cholangitis in Mice
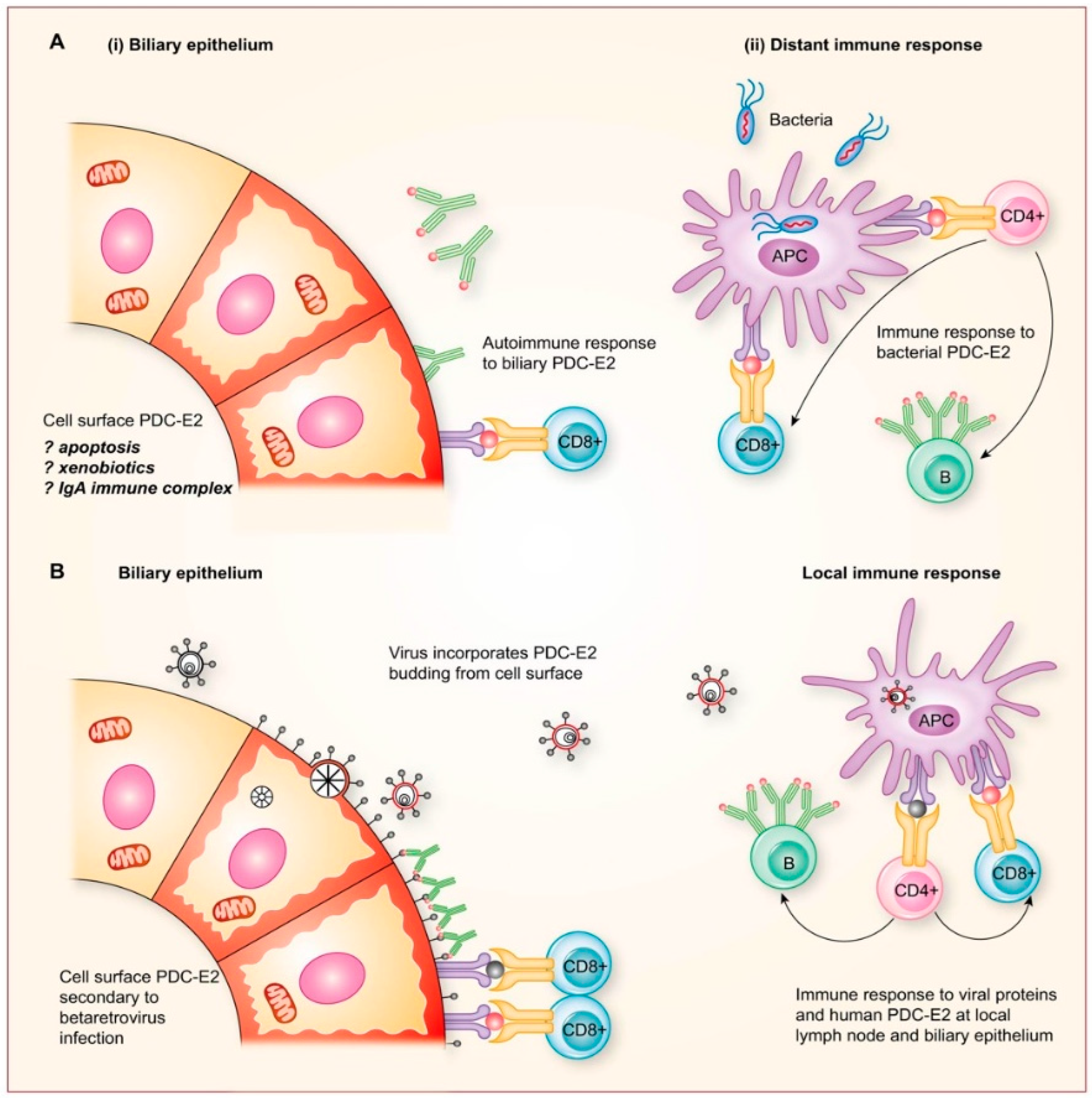
2.8. Potential Mechanisms for Betaretrovirus-Induced AMA Production
3. Modified Koch’s Postulates Linking HBRV with PBC In Vitro
- Agent must be isolated from patients with disease; HBRV has been isolated using Hs587T in co-culture with lymph node homogenates from PBC patients [44].
- Agent must cause disease when introduced to a healthy organism. HBRV and pure MMTV isolates induce the autoimmune phenotype in healthy BEC in vitro [54].
4. Bradford Hill Causal Criteria for Connecting HBRV and PBC
4.1. Strength of the Association between HBRV and PBC
- The first evidence suggestive of viral infection and the development of PBC was the detection of cross-reactive antibodies to known retroviruses in the serum of PBC patients [117]. However, the seroreactivity was non-specific and also found in patients with other cholestatic disorders, including primary sclerosing cholangitis and biliary atresia. Subsequent ELISA studies using HBRV gp52 Surface protein and incorporating large populations of age-matched healthy subjects and blood donors showed that HBRV reactivity was more specific for patients with PBC and breast cancer [91].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Criteria | Link of HBRV Infection with PBC | References |
|---|---|---|
| Strength of association * |
| [69] [46] [54,114] [53,55] [54,70] [55] [55] [56,57,58,59,60] [56,57,58,59,60,66,67] |
| Specificity * |
| [6,21] [69,92] [69] [68,69,92] [92] [1,2,53,54] [53,96] [53] |
| Consistency * |
| [54,69] [54,69] [54,69,92] [46,91,118] [46,91] |
| Biological gradient |
| [55,56,57,58,59,60,66,67] [28,51] |
| Temporality |
| [28,51] |
| Plausibility Coherence * |
| [26] [119] [54,106,107] [1,2] [56,57,58,59,60] [1,2] |
| Analogy |
| [120,121,122] [6,123] [124] |
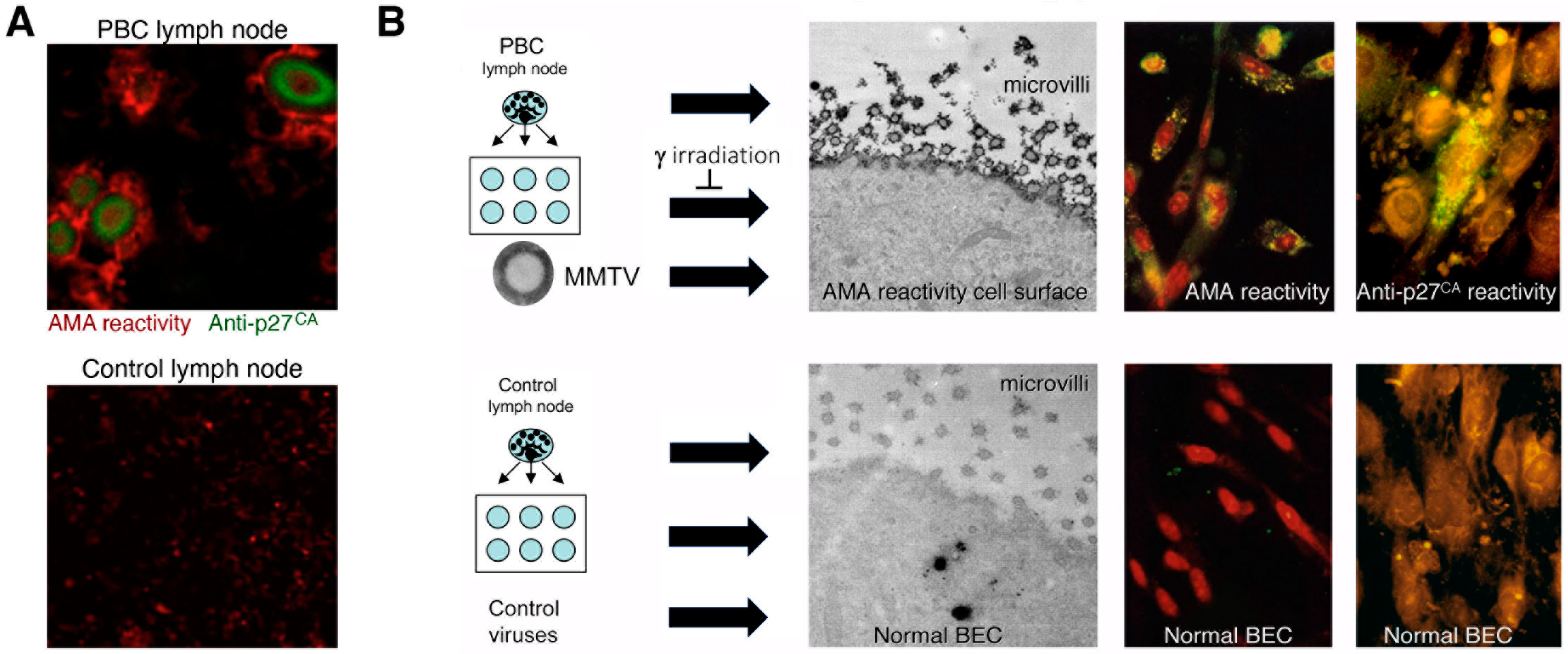
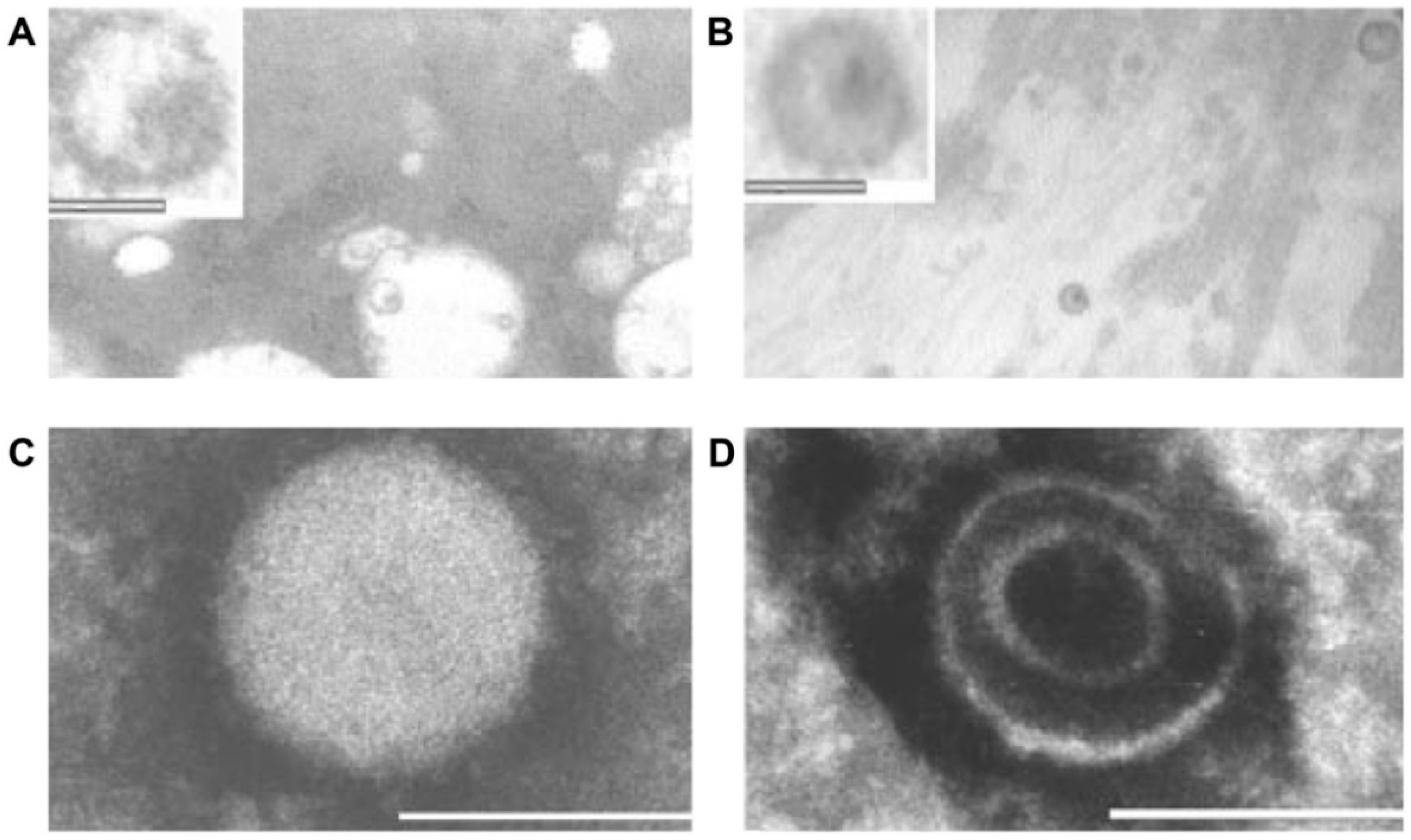
- Additional electron microscopy studies identified B-type betaretrovirus-like particles in biliary epithelium extracted from PBC patients receiving liver transplantation and also in conditioned media from PBC lymph node co-culture studies [54]. These particles were observed in all three patients with PBC, but only one particle was detected in five control patients’ biliary epithelium. Conditioned media from the co-cultivation studies also contained virus-like particles with the hydrodynamic and genomic features of HBRV and the morphological appearance of B-type particles with an electron-dense nucleocapsid adjacent to the envelope (Figure 5). Subsequently, transmissible HBRV virions have been isolated from PBC lymph nodes that display the appearance of B-type particles with an asymmetric nucleocapsid similar to MMTV, as recently reported in this Special Issue [68].
- The HBRV genome was first detected in a PBC biliary epithelium cDNA library using degenerate PCR primers capable of amplifying any retroviral pol sequence, and the sequence was very closely related to MMTV with 97–98% identity [54]. Subsequently, the proviral genome was cloned and sequenced from lymph node DNA [70], and in a recent study published in this issue, HBRV was isolated from PBC lymph nodes and sequenced [68]. The human- and mouse-derived betaretrovirus nucleotide sequences share near identity but display consistent differences expected from isolates derived from separate species [68].
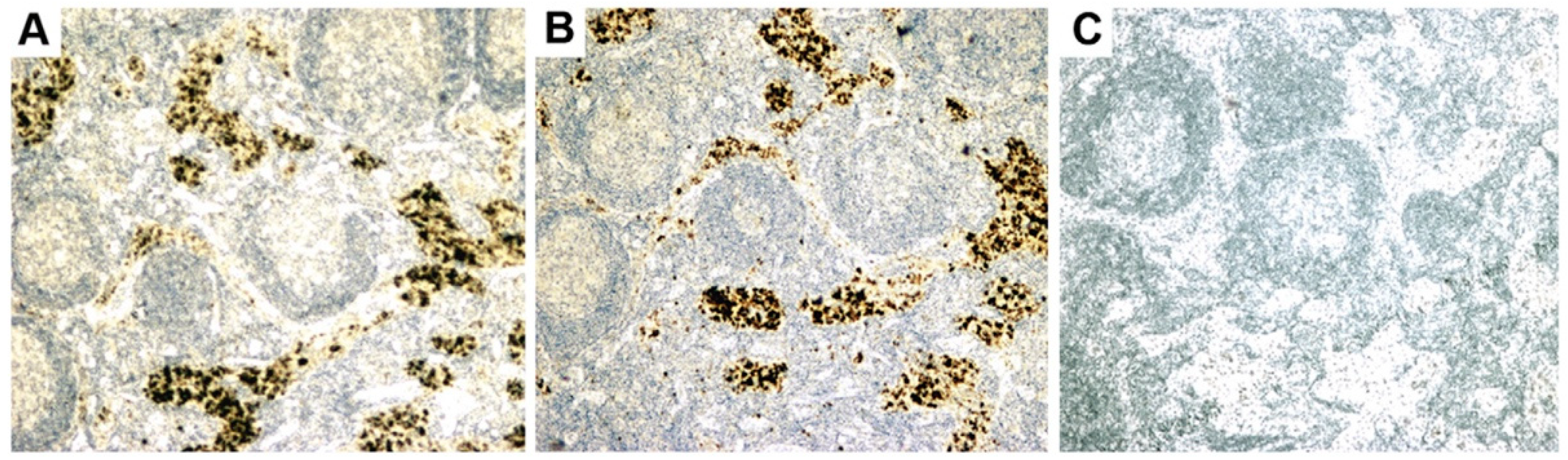
- Initial RT-PCR studies to detect HBRV RNA in the liver and perihepatic lymph node samples revealed that lymph nodes had the highest viral burden [54]. Viral proteins were easily detected by immunochemistry in PBC lymph node samples (Figure 2 and Figure 4) but not liver, where HBRV RNA was found in 73% of perihepatic lymph nodes from PBC patients and also in 20% of control patients. Liver tissue had considerably less viral burden, where 29% of the livers from PBC patients were positive for viral cDNA, compared with 7% of control patient livers [54].
- Selmi and colleagues suggested “In our opinion, the only possible final evidence for a role of a betaretrovirus in PBC could be provided by the direct demonstration of the insertion of viral sequences in the genome of a large number of patients with PBC” [112]. As the gold standard for confirming retroviral infection is the detection of proviral integrations in the genome, we used ligation-mediated PCR to identify insertions of HBRV in the human genome [69]. We also employed next-generation sequencing to improve the sensitivity of detection and screened the sequences using a stringent pipeline to avoid picking up endogenous retroviruses, false positives from contaminated laboratory reagents, duplicates, or other problematic sequences. After screening, 58% of PBC patients were found to have HBRV integrations in their cholangiocytes, compared with only 7% of control patients. HBRV RNA was detected in the biliary epithelium of 58% of the PBC patients and in 15% of the control patients using a QuantiGene hybridization method, and 75% of the PBC patients versus 13% of the control cholangiocytes using in situ hybridization. Taken together, these results provide robust evidence for infection with an exogenous HBRV virus in PBC patients, with a higher number of retroviral integrations in BEC compared with the liver [69].
- Another line of evidence for the involvement of HBRV in PBC comes from serological studies. Using MMTV isolates, Selmi and colleagues were unable to detect any serological reactivity in PBC patients’ serum [112]. To create a more suitable assay, we expressed HBRV gp52 surface protein in human cells and used the soluble HBRV gp52 to create an ELISA assay. We observed HBRV gp52 antibody response in sera from 11.5% of the PBC patients, which was similar to that detected in breast cancer patients (10.2%); blood donors and age-matched control subjects had a 2–3% seroprevalence [91]. In total, these studies suggest that HBRV may be quite prevalent in the general population.
- As the proportion of PBC patients with viral integrations exceeded the seroprevalence of anti-HBRV gp52 surface reactivity, we performed further studies to determine whether HBRV may inhibit immune responses. Retroviruses may harbor immunosuppressive domains in their envelope protein that inhibit immune responses by stimulating the regulatory cytokine, interleukin 10 [127]. By immunoscreening healthy peripheral blood mononuclear cells with overlapping 15–20mer peptides derived from the HBRV envelope protein, we characterized three immunosuppressive domains that trigger IL-10 to inhibit immune responses [128]. Notably, a similar process has been described in neonatal mice, where MMTV infection triggers IL-10 production and inhibits the production of neutralizing antibodies [82].
- To study the cellular immune responses to HBRV infection in patients with liver disease and breast cancer, we have stimulated lymphocytes with overlapping Gag and envelope peptides and used flow cytometry, ELISpot, and ELISA to measure interferon-γ release. Our preliminary studies suggest that 100% of the intrahepatic lymphocytes and 50% of the peripheral blood mononuclear cells from PBC patients make interferon-γ as compared with ~10% of liver disease controls [46].
4.2. Specificity of the Association between HBRV and PBC
4.3. Consistency of the Association between HBRV and PBC
4.4. Biological Gradient/Experiment with Response to Antiviral Therapy
4.5. Temporality
4.6. Plausibility and Coherence
4.7. Analogy
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| AMA | antimitochondrial antibodies |
| ART | antiretroviral therapy |
| BEC | biliary epithelial cells |
| HBRV | Human betaretrovirus |
| HIV | Human immunodeficiency virus |
| MMTV | mouse mammary tumor virus |
| PDC-E2 | pyruvate dehydrogenase E2 |
| LT | Liver transplantation |
| PBC | Primary biliary cholangitis |
References
- European Association for the Study of the Liver; Hirschfield, G.M.; Beuers, U.; Corpechot, C.; Invernizzi, P.; Jones, D.; Marzioni, M.; Schramm, C. EASL Clinical Practice Guidelines: The diagnosis and management of patients with primary biliary cholangitis. J. Hepatol. 2017, 67, 145–172. [Google Scholar] [CrossRef] [PubMed]
- Lindor, K.D.; Bowlus, C.L.; Boyer, J.; Levy, C.; Mayo, M. Primary Biliary Cholangitis: 2018 Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology 2019, 69, 394–419. [Google Scholar] [PubMed]
- Tanaka, A.; Leung, P.S.C.; Gershwin, M.E. Pathogen infections and primary biliary cholangitis. Clin. Exp. Immunol. 2019, 195, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Joplin, R.; Gershwin, M.E. Ductular expression of autoantigens in primary biliary cirrhosis. Semin. Liver Dis. 1997, 17, 97–104. [Google Scholar] [CrossRef]
- Joplin, R.E.; Wallace, L.L.; Lindsay, J.G.; Palmer, J.M.; Yeaman, S.J.; Neuberger, J.M. The human biliary epithelial cell plasma membrane antigen in primary biliary cirrhosis: Pyruvate dehydrogenase X? Gastroenterology 1997, 113, 1727–1733. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Chapman, R.W.; Karlsen, T.H.; Lammert, F.; Lazaridis, K.N.; Mason, A.L. The genetics of complex cholestatic disorders. Gastroenterology 2013, 144, 1357–1374. [Google Scholar] [CrossRef]
- Mason, A.L. Is PBC a viral infectious disease? Best Pract. Res. Clin. Gastroenterol. 2018, 34–35, 27–39. [Google Scholar] [CrossRef]
- Roberts, S.B.; Hirschfield, G.M.; Worobetz, L.J.; Vincent, C.; Flemming, J.A.; Cheung, A.; Qumosani, K.; Swain, M.; Grbic, D.; Ko, H.H.; et al. Ethnicity, disease severity, and survival in Canadian patients with primary biliary cholangitis. Hepatology 2022, 76, 303–316. [Google Scholar] [CrossRef]
- Arbour, L.; Rupps, R.; Field, L.; Ross, P.; Erikson, A.; Henderson, H.; Hill, W.; Yoshida, E. Characteristics of primary biliary cirrhosis in British Columbia’s First Nations population. Can. J. Gastroenterol. 2005, 19, 305–310. [Google Scholar] [CrossRef]
- Yoshida, E.M.; Riley, M.; Arbour, L.T. Autoimmune liver disease and the Canadian First Nations Aboriginal Communities of British Columbia’s Pacific Northwest. World J. Gastroenterol. 2006, 12, 3625–3627. [Google Scholar] [CrossRef]
- Mason, A.L.; Siminovitch, K.A. New perspectives on the complexity of genetic predisposition to autoimmune liver disease in indigenous Canadians. Liver Int. 2018, 38, 789–791. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, E.M.; Mason, A.; Peltekian, K.M.; Shah, H.; Thiele, S.; Borrelli, R.; Fischer, A. Epidemiology and liver transplantation burden of primary biliary cholangitis: A retrospective cohort study. CMAJ Open 2018, 6, E664–E670. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rutherford, G.; Manning, P.; Newton, J.L. Understanding Muscle Dysfunction in Chronic Fatigue Syndrome. J. Aging Res. 2016, 2016, 2497348. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, L.; Jones, D.E. Pathogenesis of primary biliary cirrhosis and its fatigue. Dig. Dis. 2014, 32, 615–625. [Google Scholar] [CrossRef]
- Hollingsworth, K.G.; Newton, J.L.; Robinson, L.; Taylor, R.; Blamire, A.M.; Jones, D.E. Loss of capacity to recover from acidosis in repeat exercise is strongly associated with fatigue in primary biliary cirrhosis. J. Hepatol. 2010, 53, 155–161. [Google Scholar] [CrossRef]
- Pells, G.; Mells, G.F.; Carbone, M.; Newton, J.L.; Bathgate, A.J.; Burroughs, A.K.; Heneghan, M.A.; Neuberger, J.M.; Day, D.B.; Ducker, S.J.; et al. The impact of liver transplantation on the phenotype of primary biliary cirrhosis patients in the UK-PBC cohort. J. Hepatol. 2013, 59, 67–73. [Google Scholar] [CrossRef]
- Carbone, M.; Bufton, S.; Monaco, A.; Griffiths, L.; Jones, D.E.; Neuberger, J.M. The effect of liver transplantation on fatigue in patients with primary biliary cirrhosis: A prospective study. J. Hepatol. 2013, 59, 490–494. [Google Scholar] [CrossRef]
- Manns, M.P.; Bremm, A.; Schneider, P.M.; Notghi, A.; Gerken, G.; Prager-Eberle, M.; Stradmann-Bellinghausen, B.; Meyer zum Buschenfelde, K.H.; Rittner, C. HLA DRw8 and complement C4 deficiency as risk factors in primary biliary cirrhosis. Gastroenterology 1991, 101, 1367–1373. [Google Scholar] [CrossRef]
- Vergani, D.; Wells, L.; Larcher, V.F.; Nasaruddin, B.A.; Davies, E.T.; Mieli-Vergani, G.; Mowat, A.P. Genetically determined low C4: A predisposing factor to autoimmune chronic active hepatitis. Lancet 1985, 2, 294–298. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Liu, X.; Han, Y.; Gorlov, I.P.; Lu, Y.; Xu, C.; Chen, W.; Juran, B.D.; Coltescu, C.; Mason, A.L.; et al. Variants at IRF5-TNPO3, 17q12-21 and MMEL1 are associated with primary biliary cirrhosis. Nat. Genet. 2010, 42, 655–657. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Liu, X.; Xu, C.; Lu, Y.; Xie, G.; Gu, X.; Walker, E.J.; Jing, K.; Juran, B.D.; Mason, A.L.; et al. Primary biliary cirrhosis associated with HLA, IL12A, and IL12RB2 variants. N. Engl. J. Med. 2009, 360, 2544–2555. [Google Scholar] [CrossRef] [PubMed]
- Hirschfield, G.M.; Xie, G.; Lu, E.; Sun, Y.; Juran, B.D.; Chellappa, V.; Coltescu, C.; Mason, A.L.; Milkiewicz, P.; Myers, R.P.; et al. Association of primary biliary cirrhosis with variants in the CLEC16A, SOCS1, SPIB and SIAE immunomodulatory genes. Genes Immun. 2012, 13, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Invernizzi, P.; Lu, Y.; Kosoy, R.; Lu, Y.; Bianchi, I.; Podda, M.; Xu, C.; Xie, G.; Macciardi, F.; et al. Genome-wide meta-analyses identify three loci associated with primary biliary cirrhosis. Nat. Genet. 2010, 42, 658–660. [Google Scholar] [CrossRef]
- Li, P.; Lu, G.; Cui, Y.; Wu, Z.; Chen, S.; Li, J.; Wen, X.; Zhang, H.; Mu, S.; Zhang, F.; et al. Association of IL12A Expression Quantitative Trait Loci (eQTL) with Primary Biliary Cirrhosis in a Chinese Han Population. Medicine 2016, 95, e3665. [Google Scholar] [CrossRef] [PubMed]
- Hirschfield, G.M.; Gershwin, M.E.; Strauss, R.; Mayo, M.J.; Levy, C.; Zou, B.; Selmi, C.; Marschall, H.; Lindor, K. Phase 2 study evaluating the efficacy and safety of ustekinumab in patients with primary biliary cirrhosis who had an inadequate response to ursodeoxycholic acid. J. Hepatol. 2014, 60, S189–S190. [Google Scholar] [CrossRef]
- Gershwin, M.E.; Selmi, C.; Worman, H.J.; Gold, E.B.; Watnik, M.; Utts, J.; Lindor, K.D.; Kaplan, M.M.; Vierling, J.M. Risk factors and comorbidities in primary biliary cirrhosis: A controlled interview-based study of 1032 patients. Hepatology 2005, 42, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Yang, Z.; Zhong, R. Primary biliary cirrhosis and cancer risk: A systematic review and meta-analysis. Hepatology 2012, 56, 1409–1417. [Google Scholar] [CrossRef] [PubMed]
- Montano-Loza, A.J.; Hansen, B.E.; Corpechot, C.; Roccarina, D.; Thorburn, D.; Trivedi, P.; Hirschfield, G.; McDowell, P.; Poupon, R.; Dumortier, J.; et al. Factors Associated With Recurrence of Primary Biliary Cholangitis After Liver Transplantation and Effects on Graft and Patient Survival. Gastroenterology 2019, 156, 96–107.e1. [Google Scholar] [CrossRef]
- Li, Y.Q.; Mason, A.L. Environmental Exposures and Risks in Autoimmune Liver Diseases, 1st ed.; Wiley: Hoboken, NJ, USA, 2020. [Google Scholar]
- Abdulkarim, A.S.; Petrovic, L.M.; Kim, W.R.; Angulo, P.; Lloyd, R.V.; Lindor, K.D. Primary biliary cirrhosis: An infectious disease caused by Chlamydia pneumoniae? J. Hepatol. 2004, 40, 380–384. [Google Scholar] [CrossRef]
- Long, S.A.; Quan, C.; Van de Water, J.; Nantz, M.H.; Kurth, M.J.; Barsky, D.; Colvin, M.E.; Lam, K.S.; Coppel, R.L.; Ansari, A.; et al. Immunoreactivity of organic mimeotopes of the E2 component of pyruvate dehydrogenase: Connecting xenobiotics with primary biliary cirrhosis. J. Immunol. 2001, 167, 2956–2963. [Google Scholar] [CrossRef]
- Tanaka, A.; Prindiville, T.P.; Gish, R.; Solnick, J.V.; Coppel, R.L.; Keeffe, E.B.; Ansari, A.; Gershwin, M.E. Are infectious agents involved in primary biliary cirrhosis? A PCR approach. J. Hepatol. 1999, 31, 664–671. [Google Scholar] [CrossRef]
- Selmi, C.; Balkwill, D.L.; Invernizzi, P.; Ansari, A.A.; Coppel, R.L.; Podda, M.; Leung, P.S.; Kenny, T.P.; Van De Water, J.; Nantz, M.H.; et al. Patients with primary biliary cirrhosis react against a ubiquitous xenobiotic-metabolizing bacterium. Hepatology 2003, 38, 1250–1257. [Google Scholar] [CrossRef] [PubMed]
- Mattner, J. Impact of Microbes on the Pathogenesis of Primary Biliary Cirrhosis (PBC) and Primary Sclerosing Cholangitis (PSC). Int. J. Mol. Sci. 2016, 17, 1864. [Google Scholar] [CrossRef]
- Ala, A.; Stanca, C.M.; Bu-Ghanim, M.; Ahmado, I.; Branch, A.D.; Schiano, T.D.; Odin, J.A.; Bach, N. Increased prevalence of primary biliary cirrhosis near Superfund toxic waste sites. Hepatology 2006, 43, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Lian, Z.-X.; Leung, P.S.C.; Moritoki, Y.; Tsuneyama, K.; Kurth, M.J.; Lam, K.S.; Yoshida, K.; Yang, G.-X.; Hibi, T.; et al. Loss of tolerance in C57BL/6 mice to the autoantigen E2 subunit of pyruvate dehydrogenase by a xenobiotic with ensuing biliary ductular disease. Hepatology 2008, 48, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Yoshida, K.; Leung, P.S.C.; Moritoki, Y.; Yang, G.X.; Tsuneyama, K.; Lian, Z.X.; Hibi, T.; Ansari, A.A.; Wicker, L.S.; et al. Induction of autoimmune cholangitis in non-obese diabetic (NOD).1101 mice following a chemical xenobiotic immunization. Clin. Exp. Immunol. 2009, 155, 577–586. [Google Scholar] [CrossRef]
- Gershwin, M.E.; Coppel, R.L.; Mackay, I.R. Primary biliary cirrhosis and mitochondrial autoantigens. Insights from molecular biology. Hepatology 1988, 8, 147–151. [Google Scholar] [CrossRef]
- Kaplan, M.M.; Gershwin, M.E. Primary biliary cirrhosis. N. Engl. J. Med. 2005, 353, 1261–1273. [Google Scholar] [CrossRef]
- Perrillo, R.P.; Mason, A.L.; Jacob, S.; Gerber, M.A. Hepatitis and cholestasis in a middle-aged woman [clinical conference]. Hepatology 1996, 24, 730–734. [Google Scholar] [CrossRef]
- Van-de-Water, J.; Cooper, A.; Surh, C.D.; Coppel, R.; Danner, D.; Ansari, A.; Dickson, R.; Gerswhin, M.E. Detection of autoantibodies to the recombinant 74 Kd and 52 Kd mitochondrial autoantigens of primary biliary cirrhosis. N. Engl. J. Med. 1989, 320, 1377–1380. [Google Scholar] [CrossRef]
- Krams, S.M.; Dorshkind, K.; Gershwin, M.E. Generation of biliary lesions after transfer of human lymphocytes into severe combined immunodeficient (SCID) mice. J. Exp. Med. 1989, 170, 1919–1930. [Google Scholar] [CrossRef] [PubMed]
- Krams, S.M.; Surh, C.D.; Coppel, R.L.; Ansari, A.; Ruebner, B.; Gershwin, M.E. Immunization of experimental animals with dihydrolipoamide acetyltransferase, as a purified recombinant polypeptide, generates mitochondrial antibodies but not primary biliary cirrhosis. Hepatology 1989, 9, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Neuberger, J.; Thompson, R. PBC and AMA—What is the connection? Hepatology 1999, 29, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, S.; Van de Water, J.; Ansari, A.; Nakamura, M.; Ishibashi, H.; Coppel, R.L.; Lake, J.; Keeffe, E.B.; Roche, T.E.; Gershwin, M.E. Identification and precursor frequency analysis of a common T cell epitope motif in mitochondrial autoantigens in primary biliary cirrhosis. J. Clin. Investig. 1998, 102, 1831–1840. [Google Scholar] [CrossRef]
- Abofayed, H.; Dutra, B.; Syed, H.; Rahbari, M.; Mason, A. Cellular Immune Responses to Human Betaretrovirus in Patients with Primary Biliary Cholangitis. In Proceedings of the EASL International Liver Congress, Online, 23–26 June 2021. [Google Scholar]
- Missale, G.; Bertoni, R.; Lamonaca, V.; Valli, A.; Massari, M.; Mori, C.; Rumi, M.G.; Houghton, M.; Fiaccadori, F.; Ferrari, C. Different clinical behaviors of acute hepatitis C virus infection are associated with different vigor of the anti-viral cell-mediated immune response. J. Clin. Investig. 1996, 98, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.K.; Dudley, D.D.; Afdhal, N.H.; Dienstag, J.; Rice, C.M.; Wang, L.; Houghton, M.; Walker, B.D.; Koziel, M.J. Liver-derived CTL in hepatitis C virus infection: Breadth and specificity of responses in a cohort of persons with chronic infection. J. Immunol. 1998, 160, 1479–1488. [Google Scholar]
- Dyson, J.K.; Hirschfield, G.M.; Adams, D.H.; Beuers, U.; Mann, D.A.; Lindor, K.D.; Jones, D.E. Novel therapeutic targets in primary biliary cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 147–158. [Google Scholar] [CrossRef]
- Gish, R.G.; Mason, A. Autoimmune liver disease. Current standards, future directions. Clin. Liver Dis. 2001, 5, 287–314. [Google Scholar] [CrossRef]
- Montano-Loza, A.J.; Wasilenko, S.; Bintner, J.; Mason, A.L. Cyclosporine A Protects Against Primary Biliary Cirrhosis Recurrence After Liver Transplantation. Am. J. Transpl. 2010, 10, 852–858. [Google Scholar] [CrossRef]
- Katsumi, T.; Tomita, K.; Leung, P.S.; Yang, G.X.; Gershwin, M.E.; Ueno, Y. Animal models of primary biliary cirrhosis. Clin. Rev. Allergy Immunol. 2015, 48, 142–153. [Google Scholar] [CrossRef]
- Zhang, G.; Chen, M.; Graham, D.; Subsin, B.; McDougall, C.; Gilady, S.; Kneteman, M.; Law, L.; Swain, M.; Trauner, M.; et al. Mouse mammary tumor virus in anti-mitochondrial antibody producing mouse models. J. Hepatol. 2011, 55, 876–884. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Shen, Z.; Guo, L.; Fodera, B.; Keogh, A.; Joplin, R.; O’Donnell, B.; Aitken, J.; Carman, W.; Neuberger, J.; et al. Does a betaretrovirus infection trigger primary biliary cirrhosis? Proc. Natl. Acad. Sci. USA 2003, 100, 8454–8459. [Google Scholar] [CrossRef] [PubMed]
- Sharon, D.; Chen, M.; Zhang, G.; Girgis, S.; Sis, B.; Graham, D.; McDougall, C.; Wasilenko, S.T.; Montano-Loza, A.; Mason, A.L. Impact of combination antiretroviral therapy in the NOD.c3c4 mouse model of autoimmune biliary disease. Liver Int. 2015, 35, 1442–1450. [Google Scholar] [CrossRef] [PubMed]
- Lytvyak, E.; Hosamani, I.; Montano-Loza, A.J.; Saxinger, L.; Mason, A.L. Randomized clinical trial: Combination antiretroviral therapy with tenofovir-emtricitabine and lopinavir-ritonavir in patients with primary biliary cholangitis. Can. Liver J. 2019, 2, 31–44. [Google Scholar] [CrossRef]
- Lytvyak, E.; Montano-Loza, A.; Saxinger, L.; Mason, A. Combination Anti-Retroviral Therapy Provides Reduction in Human Betaretrovirus Load and Durable Biochemical Responses in Patients with Primary Biliary Cirrhosis. Hepatology 2015, 62, 528A. [Google Scholar]
- Lytvyak, E.; Niazi, M.; Pai, R.; He, D.; Zhang, G.; Hubscher, S.G.; Mason, A.L. Combination antiretroviral therapy improves recurrent primary biliary cholangitis following liver transplantation. Liver Int. 2021, 41, 1879–1883. [Google Scholar] [CrossRef]
- Turvey, S.; Saxinger, L.; Mason, A. Apples to Apples? A Comparison of Real-World Tolerability of Antiretrovirals in Patients with Human Immunodeficiency Virus Infection and Patients with Primary Biliary Cholangitis. Viruses 2022, 14, 516. [Google Scholar] [CrossRef]
- Schembri, G.; Schober, P. Killing two birds with one stone. Lancet 2011, 377, 96. [Google Scholar] [CrossRef]
- Kanegane, H. Inflammatory bowel diseases and primary immunodeficiency diseases. Immunol. Med. 2018, 41, 154–161. [Google Scholar] [CrossRef]
- Schlesinger, M.; Benbassat, C.; Shoenfeld, Y. Complement profile in primary biliary cirrhosis. Immunol. Res. 1992, 11, 98–103. [Google Scholar] [CrossRef]
- James, S.P.; Jones, E.A.; Hoofnagle, J.H.; Strober, W. Circulating activated B cells in primary biliary cirrhosis. J. Clin. Immunol. 1985, 5, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Spengler, U.; Möller, A.; Jung, M.C.; Messer, G.; Zachoval, R.; Hoffmann, R.M.; Eisenburg, J.; Paumgartner, G.; Riethmüller, G.; Weiss, E.H.; et al. T lymphocytes from patients with primary biliary cirrhosis produce reduced amounts of lymphotoxin, tumor necrosis factor and interferon-gamma upon mitogen stimulation. J. Hepatol. 1992, 15, 129–135. [Google Scholar] [CrossRef]
- Jang, J.S.; Juran, B.D.; Cunningham, K.Y.; Gupta, V.K.; Son, Y.M.; Yang, J.D.; Ali, A.H.; Enninga, E.A.L.; Sung, J.; Lazaridis, K.N. Single-cell mass cytometry on peripheral blood identifies immune cell subsets associated with primary biliary cholangitis. Sci. Rep. 2020, 10, 12584. [Google Scholar] [CrossRef]
- Mason, A.L.; Farr, G.H.; Xu, L.; Hubscher, S.G.; Neuberger, J.M. Pilot studies of single and combination antiretroviral therapy in patients with primary biliary cirrhosis. Am. J. Gastroenterol. 2004, 99, 2348–2355. [Google Scholar] [CrossRef]
- Mason, A.L.; Lindor, K.D.; Bacon, B.R.; Vincent, C.; Neuberger, J.M.; Wasilenko, S.T. Clinical trial: Randomized controlled study of zidovudine and lamivudine for patients with primary biliary cirrhosis stabilized on ursodiol. Aliment. Pharmacol. Ther. 2008, 28, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Goubran, M.; Wang, W.; Indik, S.; Faschinger, A.; Wasilenko, S.T.; Bintner, J.; Carpenter, E.J.; Zhang, G.; Nuin, P.; Macintyre, G.; et al. Isolation of a Human Betaretrovirus from Patients with Primary Biliary Cholangitis. Viruses 2022, 14, 886. [Google Scholar] [CrossRef]
- Wang, W.; Indik, S.; Wasilenko, S.T.; Faschinger, A.; Carpenter, E.J.; Tian, Z.; Zhang, Y.; Wong, G.K.; Mason, A.L. Frequent proviral integration of the human betaretrovirus in biliary epithelium of patients with autoimmune and idiopathic liver disease. Aliment. Pharm. Ther. 2015, 41, 393–405. [Google Scholar] [CrossRef]
- Xu, L.; Sakalian, M.; Shen, Z.; Loss, G.; Neuberger, J.; Mason, A. Cloning the human betaretrovirus proviral genome from patients with primary biliary cirrhosis. Hepatology 2004, 39, 151–156. [Google Scholar] [CrossRef]
- Mason, A.L. The evidence supports a viral aetiology for primary biliary cirrhosis. J. Hepatol. 2011, 54, 1312–1314. [Google Scholar] [CrossRef]
- Lessi, F.; Grandi, N.; Mazzanti, C.M.; Civita, P.; Scatena, C.; Aretini, P.; Bandiera, P.; Fornaciari, A.; Giuffra, V.; Fornaciari, G.; et al. A human MMTV-like betaretrovirus linked to breast cancer has been present in humans at least since the copper age. Aging 2020, 12, 15978–15994. [Google Scholar] [CrossRef]
- Stewart, T.H.; Sage, R.D.; Stewart, A.F.; Cameron, D.W. Breast cancer incidence highest in the range of one species of house mouse, Mus domesticus. Br. J. Cancer 2000, 82, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.F.R.; Chen, H.H. Revisiting the MMTV Zoonotic Hypothesis to Account for Geographic Variation in Breast Cancer Incidence. Viruses 2022, 14, 559. [Google Scholar] [CrossRef] [PubMed]
- Taube, R.; Loya, S.; Avidan, O.; Perach, M.; Hizi, A. Reverse transcriptase of mouse mammary tumour virus: Expression in bacteria, purification and biochemical characterization. Biochem. J. 1998, 329 Pt 3, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Hagen, B.; Kraase, M.; Indikova, I.; Indik, S. A high rate of polymerization during synthesis of mouse mammary tumor virus DNA alleviates hypermutation by APOBEC3 proteins. PLoS Pathog. 2019, 15, e1007533. [Google Scholar] [CrossRef]
- Ross, S.R. Mouse mammary tumor virus and its interaction with the immune system. Immunol. Res. 1998, 17, 209–216. [Google Scholar] [CrossRef]
- Dudley, J.P.; Golovkina, T.V.; Ross, S.R. Lessons Learned from Mouse Mammary Tumor Virus in Animal Models. ILAR J. 2016, 57, 12–23. [Google Scholar] [CrossRef]
- Golovkina, T.V.; Chervonsky, A.; Dudley, J.P.; Ross, S.R. Transgenic mouse mammary tumor virus superantigen expression prevents viral infection. Cell 1992, 69, 637–645. [Google Scholar] [CrossRef]
- Kozma, S.; Calberg-Bacq, C.M.; Francois, C.; Osterrieth, P.M. Detection of virus antigens in Swiss albino mice infected by milk-borne mouse mammary tumour virus: The effect of age, sex and reproductive status. I. Localization by immunofluorescence of four antigens in mammary tissues and other organs. J. Gen. Virol. 1979, 45, 27–40. [Google Scholar] [CrossRef]
- Mazzanti, C.M.; Lessi, F.; Armogida, I.; Zavaglia, K.; Franceschi, S.; Al Hamad, M.; Roncella, M.; Ghilli, M.; Boldrini, A.; Aretini, P.; et al. Human saliva as route of inter-human infection for mouse mammary tumor virus. Oncotarget 2015, 6, 18355–18363. [Google Scholar] [CrossRef]
- Kane, M.; Case, L.K.; Kopaskie, K.; Kozlova, A.; MacDearmid, C.; Chervonsky, A.V.; Golovkina, T.V. Successful transmission of a retrovirus depends on the commensal microbiota. Science 2011, 334, 245–249. [Google Scholar] [CrossRef]
- Acha-Orbea, H.; Palmer, E. Mls—A retrovirus exploits the immune system [see comments]. Immunol. Today 1991, 12, 356–361. [Google Scholar] [CrossRef]
- Cotterchio, M.; Nadalin, V.; Sauer, M. Human breast cancer and lymphomas may share a common aetiology involving Mouse Mammary Tumour Virus (MMTV). Med. Hypotheses 2002, 59, 492–494. [Google Scholar] [CrossRef]
- Lawson, J.S.; Mazzanti, C.; Civita, P.; Menicagli, M.; Ngan, C.C.; Whitaker, N.J.; Hochman, J.; Braitbard, O.; Yosufi, B.; Glenn, W.K. Association of Mouse Mammary Tumor Virus with Human Breast Cancer: Histology, Immunohistochemistry and Polymerase Chain Reaction Analyses. Front. Oncol. 2018, 8, 141. [Google Scholar] [CrossRef]
- Nartey, T.; Moran, H.; Marin, T.; Arcaro, K.F.; Anderton, D.L.; Etkind, P.; Holland, J.F.; Melana, S.M.; Pogo, B.G. Human Mammary Tumor Virus (HMTV) sequences in human milk. Infect. Agents Cancer 2014, 9, 20. [Google Scholar] [CrossRef] [PubMed]
- Mason, A.L.; Gilady, S.Y.; Mackey, J.R. Mouse mammary tumor virus in human breast cancer red herring or smoking gun? Am. J. Pathol. 2011, 179, 1588–1590. [Google Scholar] [CrossRef]
- Moore, D.H.; Charney, J.; Kramarsky, B.; Lasfargues, E.Y.; Sarkar, N.H.; Brennan, M.J.; Burrows, J.H.; Sirsat, S.M.; Paymaster, J.C.; Vaidya, A.B. Search for a human breast cancer virus. Nature 1971, 229, 611–614. [Google Scholar] [CrossRef]
- Lawson, J.S.; Glenn, W.K. Mouse Mammary Tumour Virus (MMTV) in Human Breast Cancer—The Value of Bradford Hill Criteria. Viruses 2022, 14, 721. [Google Scholar] [CrossRef]
- Shao, W.; Shan, J.; Kearney, M.F.; Wu, X.; Maldarelli, F.; Mellors, J.W.; Luke, B.; Coffin, J.M.; Hughes, S.H. Retrovirus Integration Database (RID): A public database for retroviral insertion sites into host genomes. Retrovirology 2016, 13, 47. [Google Scholar] [CrossRef]
- Zhang, G.; Bashiri, K.; Kneteman, M.; Cave, K.; Hong, Y.; Mackay, J.; Alter, H.; Mason, A. Seroprevalence of Human Betaretrovirus Surface Protein Antibodies in Patients with Breast Cancer and Liver Disease. J. Oncol. 2020, 2020, 8958192. [Google Scholar]
- Johal, H.; Marie Scott, G.; Jones, R.; Camaris, C.; Riordan, S.; Rawlinson, W.D. Mouse mammary tumour virus-like virus (MMTV-LV) is present within the liver in a wide range of hepatic disorders and unrelated to nuclear p53 expression or hepatocarcinogenesis. J. Hepatol. 2009, 50, 548–554. [Google Scholar] [CrossRef]
- Prakash, O.; Mason, A.; Luftig, R.B.; Bautista, A.P. Hepatitis C virus (HCV) and human immunodeficiency virus type 1 (HIV-1) infections in alcoholics. Front. Biosci. 2002, 7, 286–300. [Google Scholar] [PubMed]
- Van-de-Water, J.; Turchany, J.; Leung, P.; Lake, J.; Munoz, S.; Surh, C.D.; Coppel, R.; Ansari, A.A.; Nakanuma, Y.; Gershwin, M.E. Molecular mimicry in primary biliary cirrhosis: Evidence for biliary epithelial expression of a molecule cross-reactive with pyruvate dehydrogenase complex E2. J. Clin. Investig. 1993, 91, 2653–2664. [Google Scholar] [CrossRef] [PubMed]
- Wasilenko, S.T.; Mason, G.E.; Mason, A.L. Primary biliary cirrhosis, bacteria and molecular mimicry: What’s the molecule and where’s the mimic? Liver Int. 2009, 29, 779–782. [Google Scholar] [CrossRef] [PubMed]
- Hohenester, S.; Beuers, U.; Medina, J.F.; Elferink, R.P. Antimitochondrial antibodies may be insufficiently specific to define primary biliary cirrhosis-like disease in mouse models. Hepatology 2013, 58, 828–830. [Google Scholar] [CrossRef]
- Irie, J.; Wu, Y.; Wicker, L.S.; Rainbow, D.; Nalesnik, M.A.; Hirsch, R.; Peterson, L.B.; Leung, P.S.; Cheng, C.; Mackay, I.R.; et al. NOD.c3c4 congenic mice develop autoimmune biliary disease that serologically and pathogenetically models human primary biliary cirrhosis. J. Exp. Med. 2006, 203, 1209–1219. [Google Scholar] [CrossRef]
- Koarada, S.; Wu, Y.; Fertig, N.; Sass, D.A.; Nalesnik, M.; Todd, J.A.; Lyons, P.A.; Fenyk-Melody, J.; Rainbow, D.B.; Wicker, L.S.; et al. Genetic control of autoimmunity: Protection from diabetes, but spontaneous autoimmune biliary disease in a nonobese diabetic congenic strain. J. Immunol. 2004, 173, 2315–2323. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Lian, Z.X.; Moritoki, Y.; Lan, R.Y.; Tsuneyama, K.; Chuang, Y.H.; Yang, G.X.; Ridgway, W.; Ueno, Y.; Ansari, A.A.; et al. IL-2 receptor alpha(−/−) mice and the development of primary biliary cirrhosis. Hepatology 2006, 44, 1240–1249. [Google Scholar] [CrossRef]
- Oertelt, S.; Lian, Z.X.; Cheng, C.M.; Chuang, Y.H.; Padgett, K.A.; He, X.S.; Ridgway, W.M.; Ansari, A.A.; Coppel, R.L.; Li, M.O.; et al. Anti-mitochondrial antibodies and primary biliary cirrhosis in TGF-beta receptor II dominant-negative mice. J. Immunol. 2006, 177, 1655–1660. [Google Scholar] [CrossRef]
- Zhang, W.; Sharma, R.; Ju, S.T.; He, X.S.; Tao, Y.; Tsuneyama, K.; Tian, Z.; Lian, Z.X.; Fu, S.M.; Gershwin, M.E. Deficiency in regulatory T cells results in development of antimitochondrial antibodies and autoimmune cholangitis. Hepatology 2009, 49, 545–552. [Google Scholar] [CrossRef]
- Montano-Loza, A.J.; Wasilenko, S.; Bintner, J.; Mason, A.L. Cyclosporine A inhibits in vitro replication of betaretrovirus associated with primary biliary cirrhosis. Liver Int. 2010, 30, 871–877. [Google Scholar] [CrossRef]
- Huang, W.; Rainbow, D.B.; Wu, Y.; Adams, D.; Shivakumar, P.; Kottyan, L.; Karns, R.; Aronow, B.; Bezerra, J.; Gershwin, M.E.; et al. A Novel Pkhd1 Mutation Interacts with the Nonobese Diabetic Genetic Background To Cause Autoimmune Cholangitis. J. Immunol. 2018, 200, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Varmus, H.E.; Padgett, T.; Heasley, S.; Simon, G.; Bishop, J.M. Cellular functions are required for the synthesis and integration of avian sarcoma virus-specific DNA. Cell 1977, 11, 307–319. [Google Scholar] [CrossRef]
- Lamb, R.; Bonuccelli, G.; Ozsvari, B.; Peiris-Pages, M.; Fiorillo, M.; Smith, D.L.; Bevilacqua, G.; Mazzanti, C.M.; McDonnell, L.A.; Naccarato, A.G.; et al. Mitochondrial mass, a new metabolic biomarker for stem-like cancer cells: Understanding WNT/FGF-driven anabolic signaling. Oncotarget 2015, 6, 30453–30471. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Willows, S.; Syed, H.; Mason, A. Metabolic Remodeling and Mitochondrial Biogenesis in Primary Biliary Cholangitis is Linked with Betaretrovirus Infection. In Canadian Liver Meeting Abstracts; University of Toronto Press: Toronto, ON, Canada, 2022. [Google Scholar]
- Wysokinski, F.; Sharon, D.; Zhang, G.; Mason, A. Relationship of Betaretroviral infection with differential expression of metabolic enzymes. Can. J. Gastroenterol. Hepatol. 2015, 29, 158A. [Google Scholar]
- Hsu, P.N.; Wolf Bryant, P.; Sutkowski, N.; McLellan, B.; Ploegh, H.L.; Huber, B.T. Association of mouse mammary tumor virus superantigen with MHC class II during biosynthesis. J. Immunol. 2001, 166, 3309–3314. [Google Scholar] [CrossRef] [PubMed]
- Fredericks, D.N.; Relman, D.A. Sequence-based identification of microbial pathogens: A reconsideration of Koch’s postulates. Clin. Microbiol. Rev. 1996, 9, 18–33. [Google Scholar] [CrossRef] [PubMed]
- Sharon, D.; Mason, A.L. Role of Novel Retroviruses in Chronic Liver Disease: Assessing the Link of Human Betaretrovirus with Primary Biliary Cirrhosis. Curr. Infect. Dis. Rep. 2015, 17, 460. [Google Scholar] [CrossRef] [PubMed]
- Colapietro, F.; Lleo, A.; Generali, E. Antimitochondrial Antibodies: From Bench to Bedside. Clin. Rev. Allergy Immunol. 2021. [Google Scholar] [CrossRef]
- Selmi, C.; Ross, S.R.; Ansari, A.A.; Invernizzi, P.; Podda, M.; Coppel, R.L.; Gershwin, M.E. Lack of immunological or molecular evidence for a role of mouse mammary tumor retrovirus in primary biliary cirrhosis. Gastroenterology 2004, 127, 493–501. [Google Scholar] [CrossRef]
- Tinmouth, J.; Lee, M.; Wanless, I.R.; Tsui, F.W.; Inman, R.; Heathcote, E.J. Apoptosis of biliary epithelial cells in primary biliary cirrhosis and primary sclerosing cholangitis. Liver 2002, 22, 228–234. [Google Scholar] [CrossRef]
- Sadamoto, T.; Joplin, R.; Keogh, A.; Mason, A.; Carman, W.; Neuberger, J. Expression of pyruvate-dehydrogenase complex PDC-E2 on biliary epithelial cells induced by lymph nodes from primary biliary cirrhosis. Lancet 1998, 352, 1595–1596. [Google Scholar] [CrossRef]
- Hill, A.B. The Environment and Disease: Association or Causation? J. R. Soc. Med. 1965, 58, 295–300. [Google Scholar] [CrossRef]
- Fedak, K.M.; Bernal, A.; Capshaw, Z.A.; Gross, S. Applying the Bradford Hill criteria in the 21st century: How data integration has changed causal inference in molecular epidemiology. Emerg. Themes Epidemiol. 2015, 12, 14. [Google Scholar] [CrossRef] [PubMed]
- Mason, A.L.; Xu, L.; Guo, L.; Munoz, S.; Jaspan, J.B.; Bryer-Ash, M.; Cao, Y.; Sander, D.M.; Shoenfeld, Y.; Ahmed, A.; et al. Detection of retroviral antibodies in primary biliary cirrhosis and other idiopathic biliary disorders. Lancet 1998, 351, 1620–1624. [Google Scholar] [CrossRef]
- Rahbari, M.; Sharon, D.; Landi, A.; Houghton, M.; Mason, A. Virological footprint of T-Cell responses in patients with primary biliary cirrhosis. Can. J. Gastroenterol. Hepatol. 2015, 29, 159A. [Google Scholar]
- Le Ricousse, S.; Gouilleux, F.; Fortin, D.; Joulin, V.; Richard-Foy, H. Glucocorticoid and progestin receptors are differently involved in the cooperation with a structural element of the mouse mammary tumor virus promoter. Proc. Natl. Acad. Sci. USA 1996, 93, 5072–5077. [Google Scholar] [CrossRef]
- Einsiedel, L.; Chiong, F.; Jersmann, H.; Taylor, G.P. Human T-cell leukaemia virus type 1 associated pulmonary disease: Clinical and pathological features of an under-recognised complication of HTLV-1 infection. Retrovirology 2021, 18, 1–13. [Google Scholar] [CrossRef]
- Nakamura, H.; Tsukamoto, M.; Nagasawa, Y.; Kitamura, N.; Shimizu, T.; Kawakami, A.; Nagata, K.; Takei, M. Does HTLV-1 Infection Show Phenotypes Found in Sjögren’s Syndrome? Viruses 2022, 14, 100. [Google Scholar] [CrossRef]
- Mason, A.; Theal, J.; Bain, V.; Adams, E.; Perrillo, R. Hepatitis B Virus Replication in Damaged Endothelial Tissues of Patients with Extrahepatic Disease. Am. J. Gastroenterol. 2005, 100, 972–976. [Google Scholar] [CrossRef]
- Lenzi, M.; Frisoni, M.; Mantovani, V.; Ricci, P.; Muratori, L.; Francesconi, R.; Cuccia, M.; Ferri, S.; Bianchi, F.B. Haplotype HLA-B8-DR3 confers susceptibility to hepatitis C virus-related mixed cryoglobulinemia. Blood 1998, 91, 2062–2066. [Google Scholar] [CrossRef]
- Lawson, J.S. Do viruses cause breast cancer? Methods Mol. Biol. 2009, 471, 421–438. [Google Scholar] [PubMed]
- Nakanuma, Y.; Kono, N.; Ohta, G.; Kato, Y.; Kobayashi, K. Ultrastructural changes of bile duct epithelium in primary biliary cirrhosis in relation to progression of bile duct loss. Virchows Arch. A 1982, 398, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Nakanuma, Y.; Ohta, G.; Kono, N.; Kobayashi, K.; Kato, Y. Electron microscopic observation of destruction of biliary epithelium in primary biliary cirrhosis. Liver 1983, 3, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Denner, J.; Eschricht, M.; Lauck, M.; Semaan, M.; Schlaermann, P.; Ryu, H.; Akyuz, L. Modulation of cytokine release and gene expression by the immunosuppressive domain of gp41 of HIV-1. PLoS ONE 2013, 8, e55199. [Google Scholar] [CrossRef]
- Rahbari, M.; Sharon, D.; Houghton, M.; Mason, A. Identification of an immunosuppressive domain in human betaretrovirus. In Proceedings of the Canadian Digestive Diseases Week (CDDW) Annual Meeting, Banff, AB, Canada, 3–6 March 2017. [Google Scholar]
- Mason, A.L.; Wasilenko, S.T. Other potential medical therapies: The use of antiviral agents to investigate and treat primary ciliary cirrhosis. Clin. Liver Dis. 2008, 12, 445–460. [Google Scholar] [CrossRef]
- Pader, J.; Basmadjian, R.B.; O’Sullivan, D.E.; Mealey, N.E.; Ruan, Y.; Friedenreich, C.; Murphy, R.; Wang, E.; Quan, M.L.; Brenner, D.R. Examining the etiology of early-onset breast cancer in the Canadian Partnership for Tomorrow’s Health (CanPath). Cancer Causes Control 2021, 32, 1117–1128. [Google Scholar] [CrossRef]
- Dong, V.; Montano-Loza, A.; Mason, A. Modelling Recurrent Primary Biliary Cholangitis and Primary Sclerosing Cholangitis as Infectious Diseases Following Liver Transplantation. OBM Transplant. 2019, 3, 94. [Google Scholar] [CrossRef]
- Van De Water, J.; Gerson, L.B.; Ferrell, L.D.; Lake, J.R.; Coppel, R.L.; Batts, K.P.; Wiesner, R.H.; Gershwin, M.E. Immunohistochemical evidence of disease recurrence after liver transplantation for primary biliary cirrhosis. Hepatology 1996, 24, 1079–1084. [Google Scholar] [CrossRef]
- Wasilenko, S.T.; Montano-Loza, A.J.; Mason, A.L. Is there a Role for Cyclophilin Inhibitors in the Management of Primary Biliary Cirrhosis? Viruses 2013, 5, 423–438. [Google Scholar] [CrossRef]
- Berenguer, M.; Prieto, M.; Cordoba, J.; Rayon, J.M.; Carrasco, D.; Olaso, V.; San-Juan, F.; Gobernado, M.; Mir, J.; Berenguer, J. Early development of chronic active hepatitis in recurrent hepatitis C virus infection after liver transplantation: Association with treatment of rejection. J. Hepatol. 1998, 28, 756–763. [Google Scholar] [CrossRef]
- Acha-Orbea, H.; Shakhov, A.N.; Finke, D. Immune response to MMTV Infection. Front. Biosci. 2007, 12, 1594–1609. [Google Scholar] [CrossRef] [PubMed]
- Buetti, E.; Diggelmann, H. Cloned mouse mammary tumor virus DNA is biologically active in transfected mouse cells and its expression is stimulated by glucocorticoid hormones. Cell 1981, 23, 335–345. [Google Scholar] [CrossRef]
- Bergasa, N.; Mason, A.; Floreani, A.; Heathcote, J.; Swain, M.; Jones, D.; Lindor, K.; Bassendine, M.; Worman, H. Primary biliary cirrhosis: Report of a focus study group. Hepatology 2004, 40, 1013–1020. [Google Scholar] [CrossRef] [PubMed]
- Ashouri, J.; McCarthy, E.; Yu, S.; Perlmutter, N.; Lin, C.; DeRisi, J.; Ye, C.J.; Weiss, A. Naïve arthritogenic SKG T cells have a defect in anergy and a repertoire pruned by superantigen. bioRxiv 2022. bioRxiv:2022.01.13.476250. [Google Scholar] [CrossRef]
- Asuri, S.; McIntosh, S.; Taylor, V.; Rokeby, A.; Kelly, J.; Shumansky, K.; Field, L.L.; Yoshida, E.M.; Arbour, L. Primary Biliary Cholangitis in British Columbia First Nations: Clinical features and discovery of novel genetic susceptibility loci. Liver Int. 2018, 38, 940–948. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Syed, H.; Penner, T.; Mason, A.L. Linking Human Betaretrovirus with Autoimmunity and Liver Disease in Patients with Primary Biliary Cholangitis. Viruses 2022, 14, 1941. https://doi.org/10.3390/v14091941
Syed H, Penner T, Mason AL. Linking Human Betaretrovirus with Autoimmunity and Liver Disease in Patients with Primary Biliary Cholangitis. Viruses. 2022; 14(9):1941. https://doi.org/10.3390/v14091941
Chicago/Turabian StyleSyed, Hussain, Tara Penner, and Andrew L. Mason. 2022. "Linking Human Betaretrovirus with Autoimmunity and Liver Disease in Patients with Primary Biliary Cholangitis" Viruses 14, no. 9: 1941. https://doi.org/10.3390/v14091941
APA StyleSyed, H., Penner, T., & Mason, A. L. (2022). Linking Human Betaretrovirus with Autoimmunity and Liver Disease in Patients with Primary Biliary Cholangitis. Viruses, 14(9), 1941. https://doi.org/10.3390/v14091941