
Assessing the Impact of SARS-CoV-2 Lineages and Mutations on Patient Survival

 ,
,  , , ,
, , ,  , , , , , , ,
, , , , , , ,  , and add
Show full author list
, and add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
2.1. Design and Patient Selection
2.2. Sequencing SARS-CoV-2 Genome
2.3. Sequencing Data Processing
2.4. Clinical Data Preprocessing
2.5. Statistical Analysis
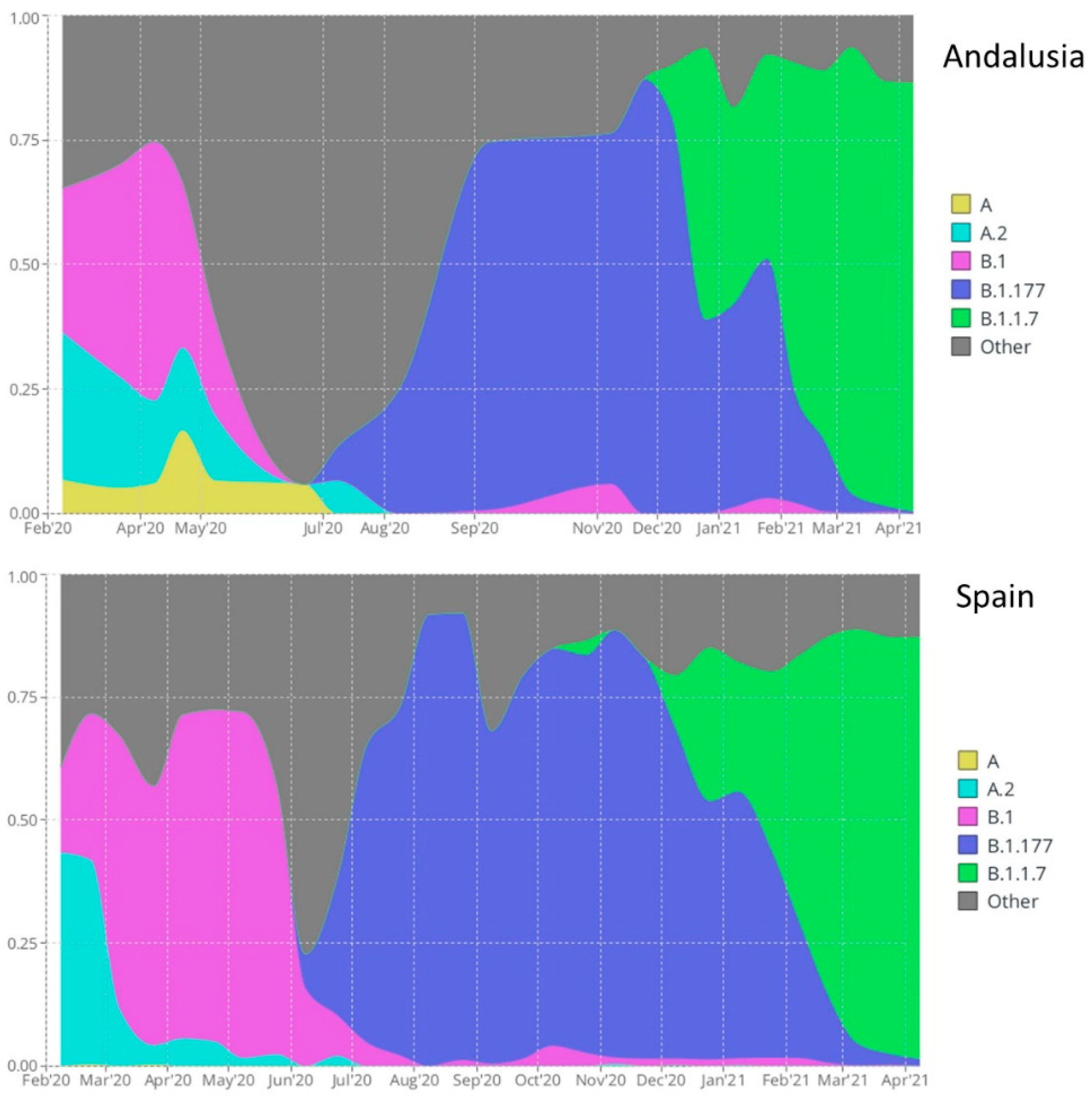
2.6. Visualization of Lineage Prevalence over Time
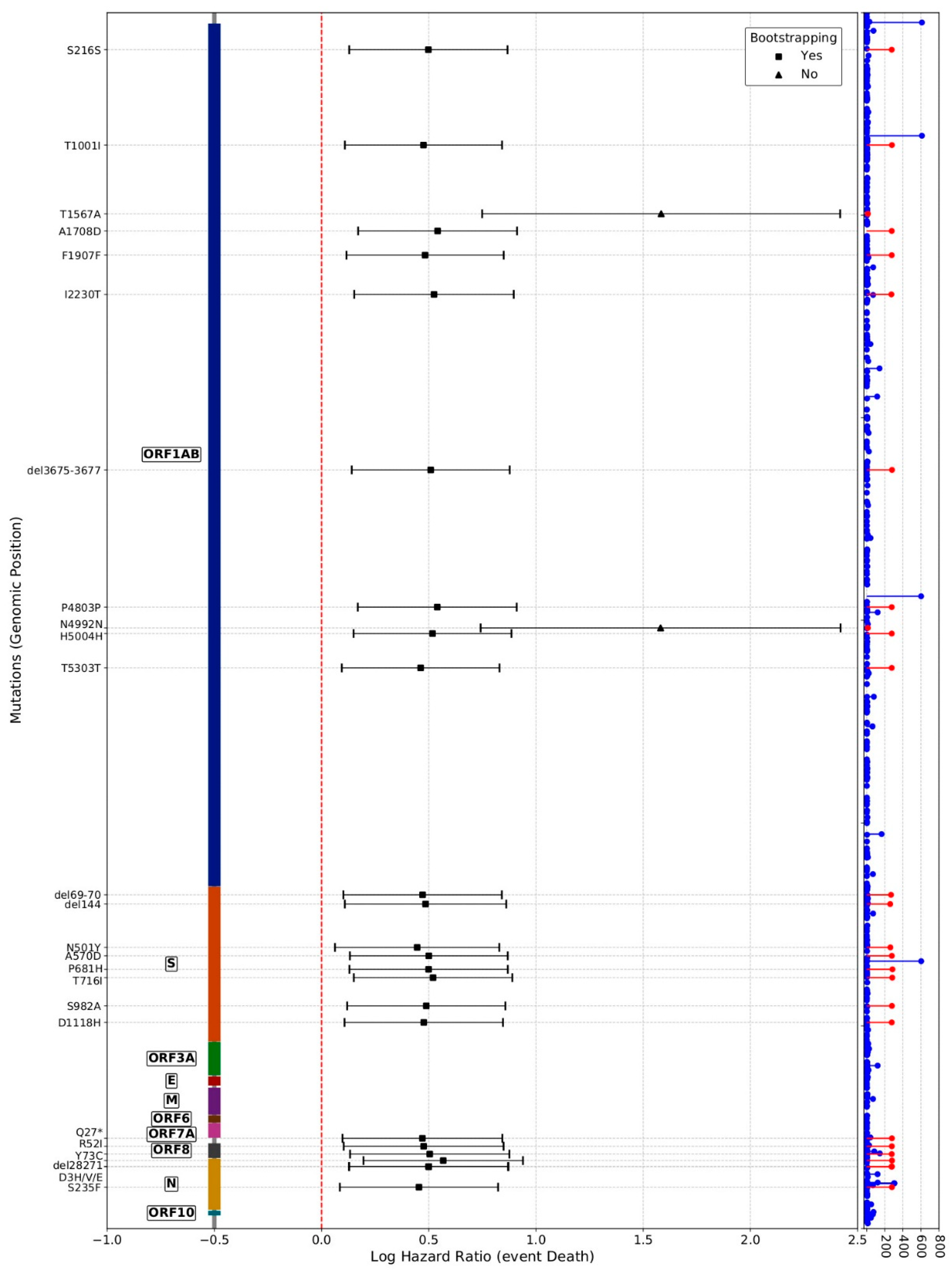
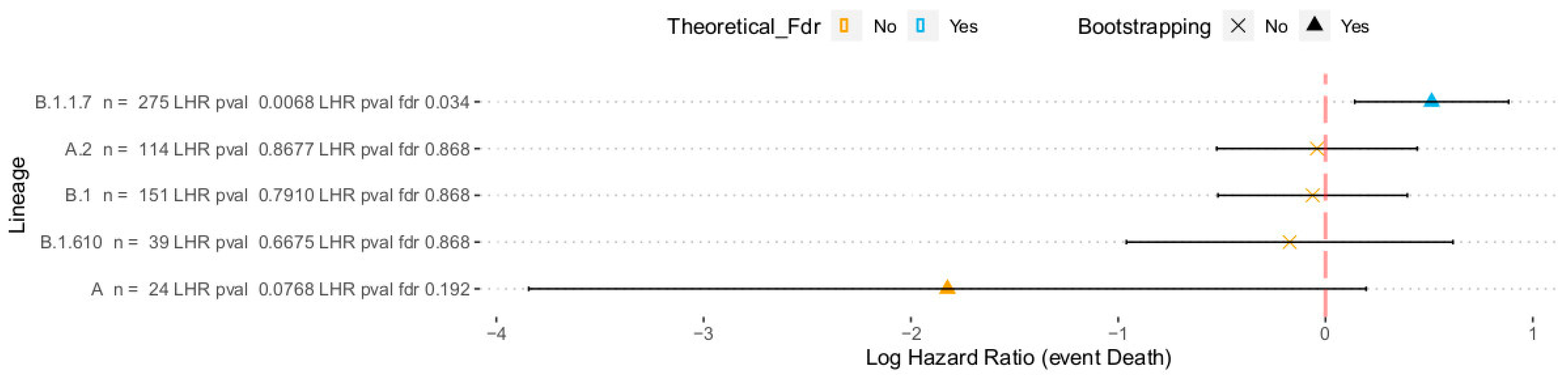
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data–from vision to reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef] [PubMed]
- Faria, N.R.; Mellan, T.A.; Whittaker, C.; Claro, I.M.; Candido, D.d.S.; Mishra, S.; Crispim, M.A.; Sales, F.C.; Hawryluk, I.; McCrone, J.T. Genomics and epidemiology of the P. 1 SARS-CoV-2 lineage in Manaus, Brazil. Science 2021, 372, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.W.; Tambyah, P.A.; Hui, D.S. Emergence of a new SARS-CoV-2 variant in the UK. J. Infect. 2021, 82, e27–e28. [Google Scholar] [CrossRef] [PubMed]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef]
- Volz, E.; Mishra, S.; Chand, M.; Barrett, J.C.; Johnson, R.; Geidelberg, L.; Hinsley, W.R.; Laydon, D.J.; Dabrera, G.; O’Toole, Á. Assessing transmissibility of SARS-CoV-2 lineage B. 1.1. 7 in England. Nature 2021, 593, 266–269. [Google Scholar] [CrossRef]
- Hodcroft, E.B.; Zuber, M.; Nadeau, S.; Vaughan, T.G.; Crawford, K.H.; Althaus, C.L.; Reichmuth, M.L.; Bowen, J.E.; Walls, A.C.; Corti, D. Spread of a SARS-CoV-2 variant through Europe in the summer of 2020. Nature 2021, 595, 707–712. [Google Scholar] [CrossRef]
- Araf, Y.; Akter, F.; Tang, Y.d.; Fatemi, R.; Parvez, M.S.A.; Zheng, C.; Hossain, M.G. Omicron variant of SARS-CoV-2: Genomics, transmissibility, and responses to current COVID--19 vaccines. J. Med. Virol. 2022, 94, 1825–1832. [Google Scholar] [CrossRef]
- Chen, R.E.; Zhang, X.; Case, J.B.; Winkler, E.S.; Liu, Y.; VanBlargan, L.A.; Liu, J.; Errico, J.M.; Xie, X.; Suryadevara, N. Resistance of SARS-CoV-2 variants to neutralization by monoclonal and serum-derived polyclonal antibodies. Nat. Med. 2021, 27, 717–726. [Google Scholar] [CrossRef]
- Beyer, D.K.; Forero, A. Mechanisms of Antiviral Immune Evasion of SARS-CoV-2. J. Mol. Biol. 2022, 434, 167265. [Google Scholar] [CrossRef]
- Cyranoski, D. Alarming COVID variants show vital role of genomic surveillance. Nature 2021, 589. [Google Scholar] [CrossRef]
- WHO. SARS-CoV-2 Variants of Concern and Variants of Interest. World Health Organization. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed on 3 August 2022).
- Kwok, A.J.; Mentzer, A.; Knight, J.C. Host genetics and infectious disease: New tools, insights and translational opportunities. Nat. Rev. Genet. 2021, 22, 137–153. [Google Scholar] [CrossRef] [PubMed]
- Fallerini, C.; Daga, S.; Mantovani, S.; Benetti, E.; Picchiotti, N.; Francisci, D.; Paciosi, F.; Schiaroli, E.; Baldassarri, M.; Fava, F. Association of Toll-like receptor 7 variants with life-threatening COVID-19 disease in males: Findings from a nested case-control study. eLife 2021, 10, e67569. [Google Scholar] [CrossRef] [PubMed]
- Severe COVID-19 GWAS Group. Genomewide association study of severe COVID-19 with respiratory failure. N. Engl. J. Med. 2020, 383, 1522–1534. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Butler-Laporte, G.; Nakanishi, T.; Morrison, D.R.; Afilalo, J.; Afilalo, M.; Laurent, L.; Pietzner, M.; Kerrison, N.; Zhao, K. A Neanderthal OAS1 isoform protects individuals of European ancestry against COVID-19 susceptibility and severity. Nat. Med. 2021, 27, 659–667. [Google Scholar] [CrossRef]
- COVID-19_Host_Genetics_Initiative. Mapping the human genetic architecture of COVID-19 by worldwide meta-analysis. Nature 2021, 600, 472–477. [Google Scholar] [CrossRef]
- Degenhardt, F.; Ellinghaus, D.; Juzenas, S.; Lerga-Jaso, J.; Wendorff, M.; Maya-Miles, D.; Uellendahl-Werth, F.; ElAbd, H.; Rühlemann, M.C.; Arora, J.; et al. Detailed stratified GWAS analysis for severe COVID-19 in four European populations. Hum. Mol. Genet. 2022. [Google Scholar] [CrossRef]
- Kousathanas, A.; Pairo-Castineira, E.; Rawlik, K.; Stuckey, A.; Odhams, C.A.; Walker, S.; Russell, C.D.; Malinauskas, T.; Millar, J.; Elliott, K.S.; et al. Whole genome sequencing identifies multiple loci for critical illness caused by COVID-19. medRxiv 2021, 2021.2009.2002.21262965. [Google Scholar] [CrossRef]
- Davies, N.G.; Jarvis, C.I.; Edmunds, W.J.; Jewell, N.P.; Diaz-Ordaz, K.; Keogh, R.H. Increased mortality in community-tested cases of SARS-CoV-2 lineage B. 1.1. 7. Nature 2021, 593, 270–274. [Google Scholar] [CrossRef]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.; Russell, T.W.; Tully, D.C.; Washburne, A.D. Estimated transmissibility and impact of SARS-CoV-2 lineage B. 1.1. 7 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef]
- Frampton, D.; Rampling, T.; Cross, A.; Bailey, H.; Heaney, J.; Byott, M.; Scott, R.; Sconza, R.; Price, J.; Margaritis, M. Genomic characteristics and clinical effect of the emergent SARS-CoV-2 B. 1.1. 7 lineage in London, UK: A whole-genome sequencing and hospital-based cohort study. Lancet Infect. Dis. 2021, 21, 1246–1256. [Google Scholar] [CrossRef]
- Toyoshima, Y.; Nemoto, K.; Matsumoto, S.; Nakamura, Y.; Kiyotani, K. SARS-CoV-2 genomic variations associated with mortality rate of COVID-19. J. Hum. Genet. 2020, 65, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Twohig, K.A.; Nyberg, T.; Zaidi, A.; Thelwall, S.; Sinnathamby, M.A.; Aliabadi, S.; Seaman, S.R.; Harris, R.J.; Hope, R.; Lopez-Bernal, J.; et al. Hospital admission and emergency care attendance risk for SARS-CoV-2 delta (B.1.617.2) compared with alpha (B.1.1.7) variants of concern: A cohort study. Lancet Infect. Dis. 2022, 22, 35–42. [Google Scholar] [CrossRef]
- Sievers, C.; Zacher, B.; Ullrich, A.; Huska, M.; Fuchs, S.; Buda, S.; Haas, W.; Diercke, M.; an der Heiden, M.; Kröger, S. SARS-CoV-2 Omicron variants BA.1 and BA.2 both show similarly reduced disease severity of COVID-19 compared to Delta, Germany, 2021 to 2022. Eurosurveillance 2022, 27, 2200396. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.; Eales, O.; Steyn, N.; Tang, D.; Bodinier, B.; Wang, H.; Elliott, J.; Whitaker, M.; Atchison, C.; Diggle, P.J.; et al. Twin peaks: The Omicron SARS-CoV-2 BA.1 and BA.2 epidemics in England. Science 2022, 376, eabq4411. [Google Scholar] [CrossRef]
- Nagy, Á.; Pongor, S.; Győrffy, B. Different mutations in SARS-CoV-2 associate with severe and mild outcome. Int. J. Antimicrob. Agents 2021, 57, 106272. [Google Scholar] [CrossRef]
- Young, B.E.; Fong, S.-W.; Chan, Y.-H.; Mak, T.-M.; Ang, L.W.; Anderson, D.E.; Lee, C.Y.-P.; Amrun, S.N.; Lee, B.; Goh, Y.S. Effects of a major deletion in the SARS-CoV-2 genome on the severity of infection and the inflammatory response: An observational cohort study. Lancet 2020, 396, 603–611. [Google Scholar] [CrossRef]
- CDC. SARS-CoV-2 Variant Classifications and Definitions. Centers for Disease Control and Prevention. Available online: https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-info.html (accessed on 3 August 2022).
- The_COVID-19_Genomics_UK_(COG-UK)_Consortium. An Integrated National Scale SARS-CoV-2 Genomic Surveillance Network. Lancet Microbe 2020, 1, e99. [Google Scholar] [CrossRef]
- Tao, K.; Tzou, P.L.; Nouhin, J.; Gupta, R.K.; de Oliveira, T.; Kosakovsky Pond, S.L.; Fera, D.; Shafer, R.W. The biological and clinical significance of emerging SARS-CoV-2 variants. Nat. Rev. Genet. 2021, 1, 1–17. [Google Scholar] [CrossRef]
- Outbreak.info. A Standardized, Open-Source Database of COVID-19 Resources and Epidemiology Data. Available online: https://outbreak.info/ (accessed on 3 August 2022).
- Zhou, D.; Dejnirattisai, W.; Supasa, P.; Liu, C.; Mentzer, A.J.; Ginn, H.M.; Zhao, Y.; Duyvesteyn, H.M.; Tuekprakhon, A.; Nutalai, R. Evidence of escape of SARS-CoV-2 variant B. 1.351 from natural and vaccine-induced sera. Cell 2021, 184, 2348–2361.e2346. [Google Scholar] [CrossRef]
- Sequencing of the SARS-CoV-2 Virus Genome for the Monitoring and Management of the COVID-19 Epidemic in Andalusia and the Rapid Generation of Prognostic and Response to Treatment Biomarkers. Available online: https://www.clinbioinfosspa.es/projects/covseq/indexEng.html (accessed on 3 August 2022).
- SARS-CoV-2 Whole Genome Sequencing Circuit in Andalusia. Available online: https://www.clinbioinfosspa.es/COVID_circuit/ (accessed on 3 August 2022).
- Dopazo, J.; Maya-Miles, D.; García, F.; Lorusso, N.; Calleja, M.Á.; Pareja, M.J.; López-Miranda, J.; Rodríguez-Baño, J.; Padillo, J.; Túnez, I. Implementing Personalized Medicine in COVID-19 in Andalusia: An Opportunity to Transform the Healthcare System. J. Pers. Med. 2021, 11, 475. [Google Scholar] [CrossRef]
- Muñoyerro-Muñiz, D.; Goicoechea-Salazar, J.; García-León, F.; Laguna-Tellez, A.; Larrocha-Mata, D.; Cardero-Rivas, M. Health record linkage: Andalusian health population database. Gac. Sanit. 2019, 34, 105–113. [Google Scholar] [CrossRef] [PubMed]
- SeqCOVID, Genomic Epidemiology of SARS-CoV-2 in Spain. Available online: https://seqcovid.csic.es/ (accessed on 21 January 2022).
- ISCIII. Integration of Genome Sequencing in the SARS-CoV-2 Surveillance (in Spanish). Available online: https://www.mscbs.gob.es/profesionales/saludPublica/ccayes/alertasActual/nCov/documentos/Integracion_de_la_secuenciacion_genomica-en_la_vigilancia_del_SARS-CoV-2.pdf (accessed on 3 August 2022).
- Artic_Network. SARS-CoV-2 Amplicon Set V3. Available online: https://artic.network/ncov-2019 (accessed on 3 August 2022).
- Patel, H.; Varona, S.; Monzón, S.; Espinosa-Carrasco, J.; Heuer, M.L.; Gabernet, G.; Julia, M.; Kelly, S.; Sameith, K.; Garcia, M. Nf-Core/Viral recon v2.4.1 - Plastered Magnesium Marmoset (2.4.1). Available online: https://doi.org/10.5281/zenodo.6320980 (accessed on 3 August 2022).
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Grubaugh, N.D.; Gangavarapu, K.; Quick, J.; Matteson, N.L.; De Jesus, J.G.; Main, B.J.; Tan, A.L.; Paul, L.M.; Brackney, D.E.; Grewal, S. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019, 20, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Picard_team Picard. A Set of Command Line Tools (in Java) for Manipulating High-Throughput Sequencing (HTS) Data and Formats Such as SAM/BAM/CRAM and VCF. Available online: http://broadinstitute.github.io/picard/ (accessed on 3 August 2022).
- Aksamentov, I.; Neher, R.A. Nextclade. Viral Genome Clade Assignment, Mutation Calling, and Sequence Quality Checks. Available online: https://github.com/nextstrain/nextclade (accessed on 3 August 2022).
- O’Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol. 2021, 7, veab064. [Google Scholar] [CrossRef]
- Huddleston, J.; Hadfield, J.; Sibley, T.R.; Lee, J.; Fay, K.; Ilcisin, M.; Harkins, E.; Bedford, T.; Neher, R.A.; Hodcroft, E.B. Augur: A bioinformatics toolkit for phylogenetic analyses of human pathogens. J. Open Source Softw. 2021, 6, 2906. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; Von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Tavaré, S. Some probabilistic and statistical problems in the analysis of DNA sequences. Lect. Math. Life Sci. 1986, 17, 57–86. [Google Scholar]
- Infrastructure for Secure Generation of Real World Evidence from Real World Data from the Andalusian Health Population Database. Available online: https://www.clinbioinfosspa.es/projects/iRWD/ (accessed on 3 August 2022).
- Sterne, J.A.; Murthy, S.; Diaz, J.V.; Slutsky, A.S.; Villar, J.; Angus, D.C.; Annane, D.; Azevedo, L.C.P.; Berwanger, O.; Cavalcanti, A.B. Association between administration of systemic corticosteroids and mortality among critically ill patients with COVID-19: A meta-analysis. JAMA J. Am. Med. Assoc. 2020, 324, 1330–1341. [Google Scholar]
- Stuart, E.A.; Lee, B.K.; Leacy, F.P. Prognostic score–based balance measures can be a useful diagnostic for propensity score methods in comparative effectiveness research. J. Clin. Epidemiol. 2013, 66, S84–S90.e81. [Google Scholar] [CrossRef]
- Hajage, D.; Chauvet, G.; Belin, L.; Lafourcade, A.; Tubach, F.; De Rycke, Y. Closed-form variance estimator for weighted propensity score estimators with survival outcome. Biom. J. 2018, 60, 1151–1163. [Google Scholar] [CrossRef]
- Austin, P.C. Variance estimation when using inverse probability of treatment weighting (IPTW) with survival analysis. Stat. Med. 2016, 35, 5642–5655. [Google Scholar] [CrossRef] [PubMed]
- Greifer, N. WeightIt: Weighting for Covariate Balance in Observational Studies. Available online: https://cran.r-project.org/package=WeightIt (accessed on 3 August 2022).
- Greifer, N. Cobalt: Covariate Balance Tables and Plots. Available online: https://cran.r-project.org/package=cobalt (accessed on 3 August 2022).
- Gutiérrez-Gutiérrez, B.; del Toro, M.D.; Borobia, A.M.; Carcas, A.; Jarrín, I.; Yllescas, M.; Ryan, P.; Pachón, J.; Carratalà, J.; Berenguer, J.; et al. Identification and validation of clinical phenotypes with prognostic implications in patients admitted to hospital with COVID-19: A multicentre cohort study. Lancet Infect. Dis. 2021, 21, 783–792. [Google Scholar] [CrossRef]
- Benjamini, Y.; Yekutieli, D. The control of false discovery rate in multiple testing under dependency. Ann. Stat. 2001, 29, 1165–1188. [Google Scholar] [CrossRef]
- CoVariants. Available online: https://covariants.org/ (accessed on 3 August 2022).
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Thorne, L.G.; Bouhaddou, M.; Reuschl, A.-K.; Zuliani-Alvarez, L.; Polacco, B.; Pelin, A.; Batra, J.; Whelan, M.V.; Hosmillo, M.; Fossati, A. Evolution of enhanced innate immune evasion by SARS-CoV-2. Nature 2022, 602, 487–495. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Lu, G.; Wang, Q.; Gao, G.F. Bat-to-human: Spike features determining ‘host jump’ of coronaviruses SARS-CoV, MERS-CoV, and beyond. Trends Microbiol. 2015, 23, 468–478. [Google Scholar] [CrossRef]
- Flower, T.G.; Buffalo, C.Z.; Hooy, R.M.; Allaire, M.; Ren, X.; Hurley, J.H. Structure of SARS-CoV-2 ORF8, a rapidly evolving immune evasion protein. Proc. Natl. Acad. Sci. USA 2021, 118, e2021785118. [Google Scholar] [CrossRef]
- Tan, Y.; Schneider, T.; Leong, M.; Aravind, L.; Zhang, D. Novel Immunoglobulin Domain Proteins Provide Insights into Evolution and Pathogenesis of SARS-CoV-2-Related Viruses. mBio 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.L.; Baric, R.S. Recombination, reservoirs, and the modular spike: Mechanisms of coronavirus cross-species transmission. J. Virol. 2010, 84, 3134–3146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangavarapu, K.; Latiff, A.A.; Mullen, J.L.; Alkuzweny, M.; Hufbauer, E.; Tsueng, G.; Haag, E.; Zeller, M.; Aceves, C.M.; Zaiets, K. Outbreak. info genomic reports: Scalable and dynamic surveillance of SARS-CoV-2 variants and mutations. medRxiv 2022, 2022.01.27.22269965. [Google Scholar] [CrossRef]
- Chen, C.; Nadeau, S.; Yared, M.; Voinov, P.; Xie, N.; Roemer, C.; Stadler, T. CoV-Spectrum: Analysis of globally shared SARS-CoV-2 data to identify and characterize new variants. Bioinformatics 2022, 38, 1735–1737. [Google Scholar] [CrossRef] [PubMed]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus Main Proteinase (3CLpro) Structure: Basis for Design of Anti-SARS Drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Zhang, B.; Jiang, X.-M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef]
- Candido, D.S.; Claro, I.M.; De Jesus, J.G.; Souza, W.M.; Moreira, F.R.; Dellicour, S.; Mellan, T.A.; Du Plessis, L.; Pereira, R.H.; Sales, F.C. Evolution and epidemic spread of SARS-CoV-2 in Brazil. Science 2020, 369, 1255–1260. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Lessells, R.J.; Giandhari, J.; Pillay, S.; Msomi, N.; Mlisana, K.; Bhiman, J.N.; von Gottberg, A.; Walaza, S. Sixteen novel lineages of SARS-CoV-2 in South Africa. Nat. Med. 2021, 27, 440–446. [Google Scholar] [CrossRef]
- Mutation N:D377Y across the SARS-CoV-2 Phylogeny. Available online: http://nextstrain.clinbioinfosspa.es/SARS-CoV-2-all?branchLabel=none>=N.377Y (accessed on 27 July 2022).


{kind=link}
{kind=link}
{kind=link}
| Code | Meaning |
|---|---|
| FECNAC | Birth date |
| FECDEF | Death date |
| SEXO | Gender |
| FEC_INGRESO | Hospital admission date |
| FEC_ALTA | Discharge date |
| MOTIVO_ALTA | Reason for the discharge: (recovery/death/admission in another hospital/voluntary discharge/retirement home/unspecified) |
| COD_PATOLOGIA_CRONICA | Hospital codes for chronic conditions |
| COD_FEC_INI_PATOLOGIA | Date of condition diagnosis |
| COD_CIE_NORMALIZADO | A mixture of ICD9 and ICD10 codes for diseases |
| DESC_CIE_NORMALIZADO | Description of the ICD |
| FECINI_DIAG | Diagnosis date |
| FECFIN_DIAG | End of the diagnosed condition |
| FUENTE_DIAG | Source of the diagnosis (hospital, emergency, etc.) |
| IND_CRONICO_HCUP | Is it a chronic disease? (yes/no) |
| Test COVID: FECHA | Test COVID date |
| Test COVID: TYPE | PCR/antigens |
| Test COVID: RESULTADO_TEST | Result of the test (positive/negative) |
| Pharmacy (Hospital and external): DESCRIPCION | List of drugs used in hospital or purchased in the pharmacies |
| Pharmacy (Hospital and external): FECHA | Dispensing date |
| VACUNA | List of vaccines |
| VACUNAFECHA | Vaccination dates |
| Mutation | Position | CDS | AAc Position | AAc Mutation | PFAM 1 | Definition | Lineages Eligible for Causal Analysis Bearing the Mutation |
|---|---|---|---|---|---|---|---|
| C3267T | 3267 | ORF1ab | 1001 | ORF1ab:T1001I | PF12379 | Betacoronavirus replicase NSP3, N-terminal | A; B.1.177; B.1.1.7 |
| A4964G | 4964 | ORF1ab | 1567 | ORF1ab:T1567A | PF08715 | Coronavirus papain-like peptidase | B.1; B.1.1.7 |
| C5388A | 5388 | ORF1ab | 1708 | ORF1ab: A1708D | PF08715 | Coronavirus papain-like peptidase | B.1; B.1.177; B.1.1.7 |
| del11288. 11297 | 11288 | ORF1ab | 3975-3677 | ORF1ab:del3675-3677 | PF08717 | Coronavirus replicase NSP8 | A; A.1; B.1; B.1.177; B.1.1.7 |
| C14676T | 14676 | ORF1ab | 4803 | ORF1ab:P4803P | PF00680 | RNA-dependent RNA polymerase | B.1; B.1.177; B.1.1.7 |
| C15279T | 15279 | ORF1ab | 5004 | ORF1ab: H5004H | PF00680 | RNA-dependent RNA polymerase | B.1; B.1.177; B.1.1.7 |
| del21766.21772 | 21766 | S | 69-70 | S:del69-70 | PF16451 | Betacoronavirus-like spike glycoprotein S1, N-terminal | A; A.1; B.1; B.1.177; B.1.1.7 |
| del21994.21997 | 21994 | S | 144 | S:Y144- | PF16451 | Betacoronavirus-like spike glycoprotein S1, N-terminal | A; A.1; B.1; B.1.177; B.1.1.7 |
| A23063T | 23063 | S | 501 | S:N501Y | PF09408 | Betacoronavirus spike glycoprotein S1, receptor binding | A; A.1; B.1; B.1.177; B.1.1.7 |
| C23271A | 23271 | S | 570 | S:A570D | PF19209 | Coronavirus spike glycoprotein S1, C-terminal | A; B.1; B.1.177; B.1.1.7 |
| C23709T | 23709 | S | 716 | S:T716I | PF01601 | Coronavirus spike glycoprotein S2 | B.1; B.1.177; B.1.1.7 |
| T24506G | 24506 | S | 982 | S:S982A | PF01601 | Coronavirus spike glycoprotein S2 | B.1; B.1.177; B.1.1.7 |
| G24914C | 24914 | S | 1118 | S:D1118H | PF01601 | Coronavirus spike glycoprotein S2 | B.1; B.1.177; B.1.1.7 |
| C27972T | 27972 | ORF8 | 27 | ORF8:Q27* | PF12093 | Betacoronavirus NS8 protein | A; B.1; B.1.177; B.1.1.7 |
| G28048T | 28048 | ORF8 | 52 | ORF8:R52I | PF12093 | Betacoronavirus NS8 protein | A; B.1; B.1.177; B.1.1.7 |
| A28111G | 28111 | ORF8 | 73 | ORF8:Y73C | PF12093 | Betacoronavirus NS8 protein | A; B.1; B.1.177; B.1.1.7 |
| C28977T | 28977 | N | 235 | N:S235F | PF00937 | Coronavirus nucleocapsid | A; B.1; B.1.177; B.1.1.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loucera, C.; Perez-Florido, J.; Casimiro-Soriguer, C.S.; Ortuño, F.M.; Carmona, R.; Bostelmann, G.; Martínez-González, L.J.; Muñoyerro-Muñiz, D.; Villegas, R.; Rodriguez-Baño, J.; et al. Assessing the Impact of SARS-CoV-2 Lineages and Mutations on Patient Survival. Viruses 2022, 14, 1893. https://doi.org/10.3390/v14091893
Loucera C, Perez-Florido J, Casimiro-Soriguer CS, Ortuño FM, Carmona R, Bostelmann G, Martínez-González LJ, Muñoyerro-Muñiz D, Villegas R, Rodriguez-Baño J, et al. Assessing the Impact of SARS-CoV-2 Lineages and Mutations on Patient Survival. Viruses. 2022; 14(9):1893. https://doi.org/10.3390/v14091893
Chicago/Turabian StyleLoucera, Carlos, Javier Perez-Florido, Carlos S. Casimiro-Soriguer, Francisco M. Ortuño, Rosario Carmona, Gerrit Bostelmann, L. Javier Martínez-González, Dolores Muñoyerro-Muñiz, Román Villegas, Jesus Rodriguez-Baño, and et al. 2022. "Assessing the Impact of SARS-CoV-2 Lineages and Mutations on Patient Survival" Viruses 14, no. 9: 1893. https://doi.org/10.3390/v14091893
APA StyleLoucera, C., Perez-Florido, J., Casimiro-Soriguer, C. S., Ortuño, F. M., Carmona, R., Bostelmann, G., Martínez-González, L. J., Muñoyerro-Muñiz, D., Villegas, R., Rodriguez-Baño, J., Romero-Gomez, M., Lorusso, N., Garcia-León, J., Navarro-Marí, J. M., Camacho-Martinez, P., Merino-Diaz, L., Salazar, A. d., Viñuela, L., The Andalusian COVID-19 Sequencing Initiative, ... Dopazo, J. (2022). Assessing the Impact of SARS-CoV-2 Lineages and Mutations on Patient Survival. Viruses, 14(9), 1893. https://doi.org/10.3390/v14091893