

Allosteric Integrase Inhibitor Influences on HIV-1 Integration and Roles of LEDGF/p75 and HDGFL2 Host Factors

 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmid DNAs and Reagents
2.2. Cells, Viruses, and Infections
2.3. Western Blotting
2.4. Preparation of Integration Site Libraries
2.5. Integration Site Determination and Mapping
2.6. Statistical Analyses
3. Results
3.1. Research Strategy
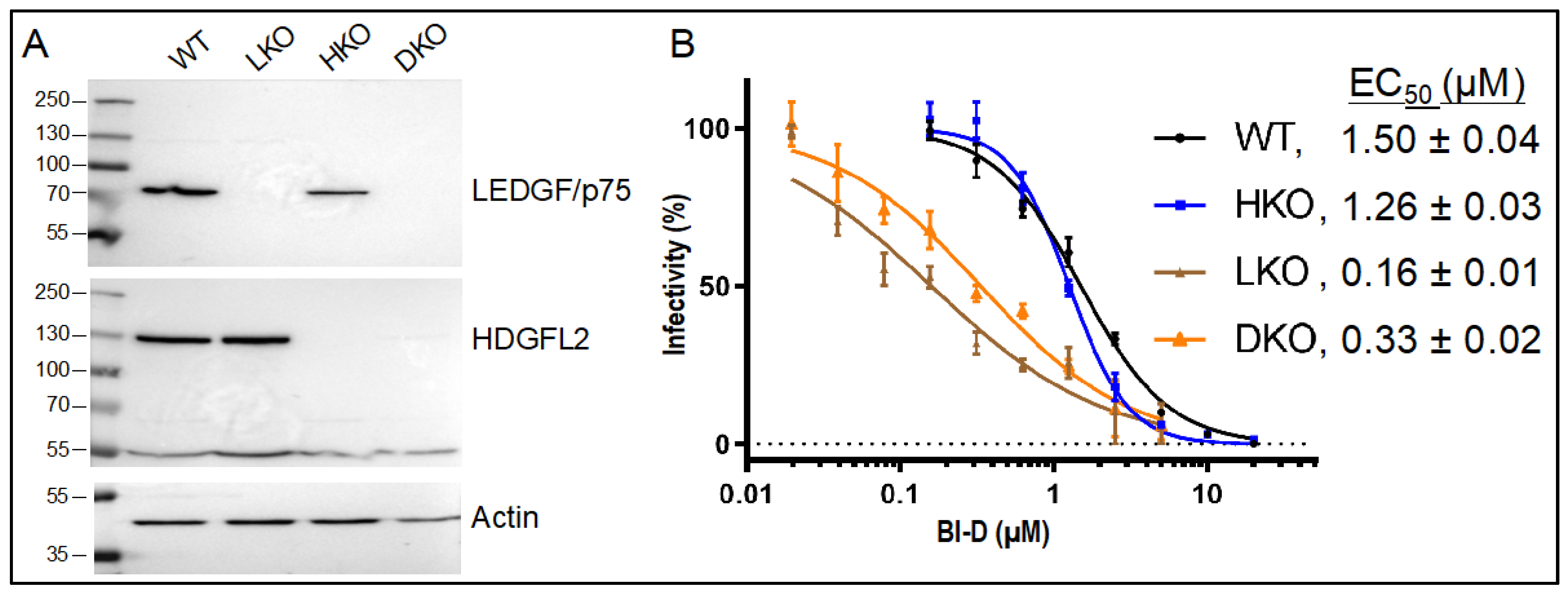
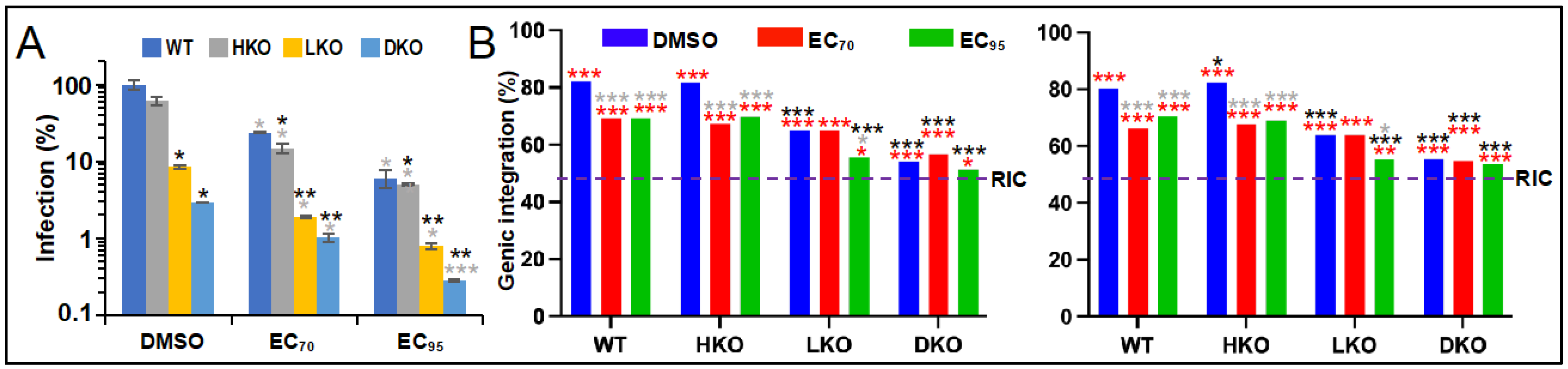
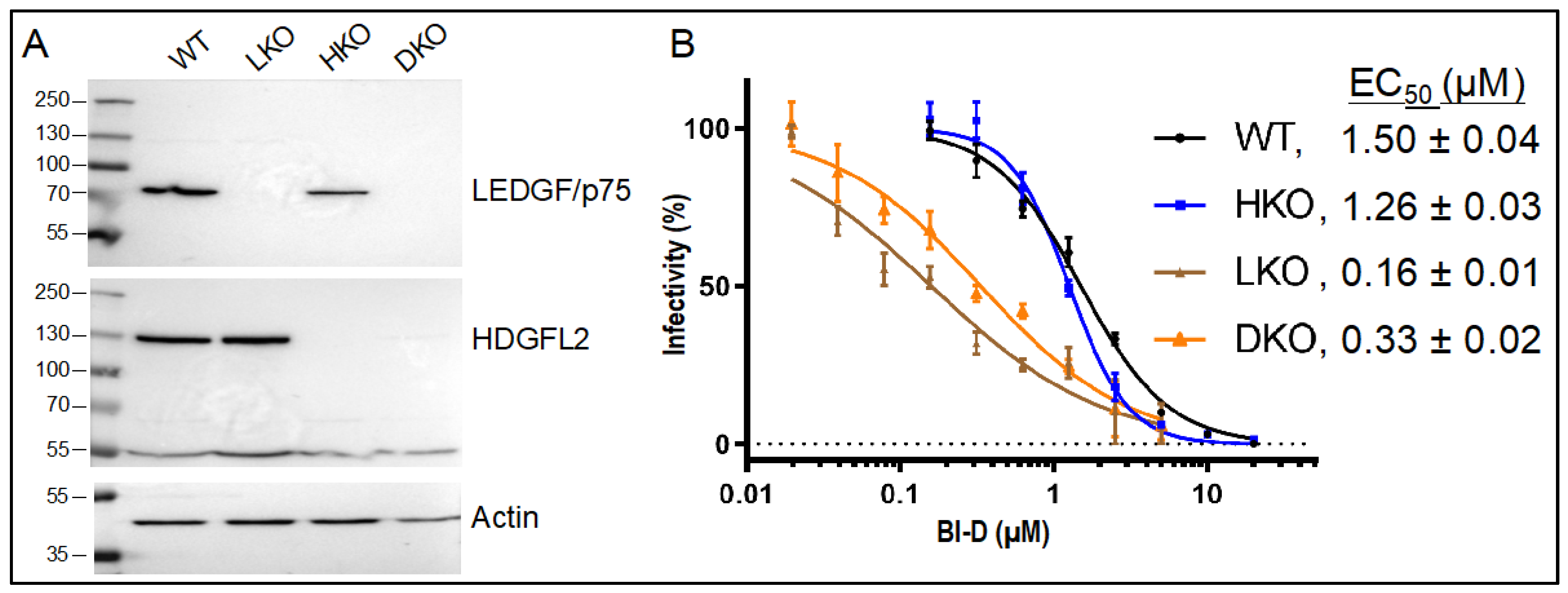
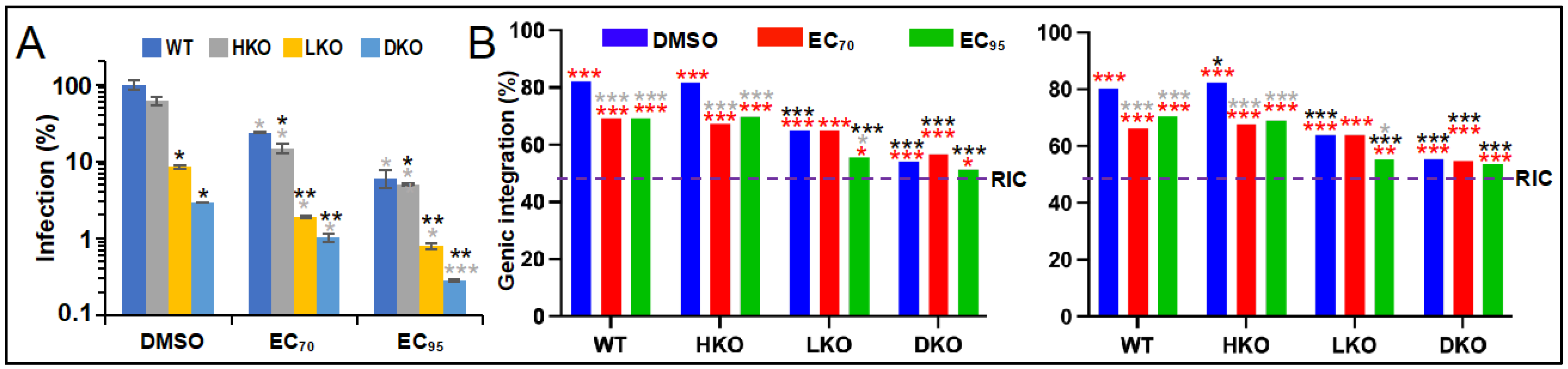
3.2. The Roles of LEDGF/p75 and HDGFL2 in ALLINI-Mediated HIV-1 Integration Retargeting
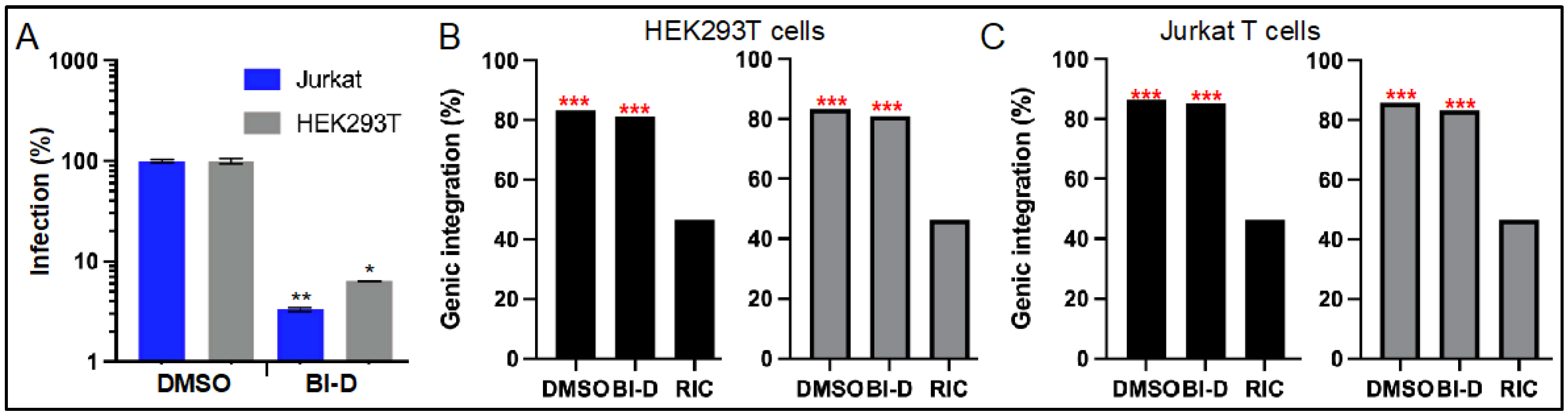
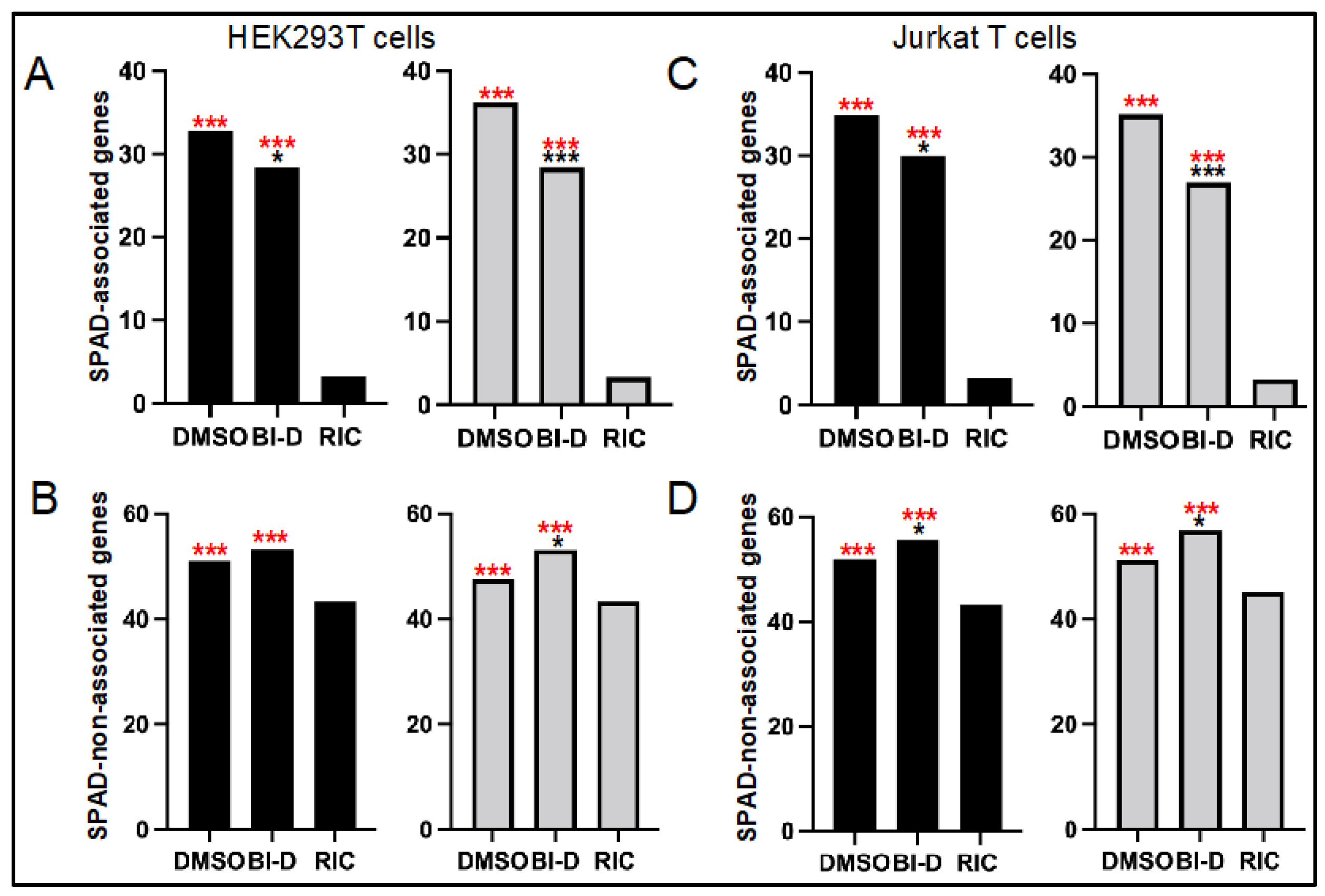
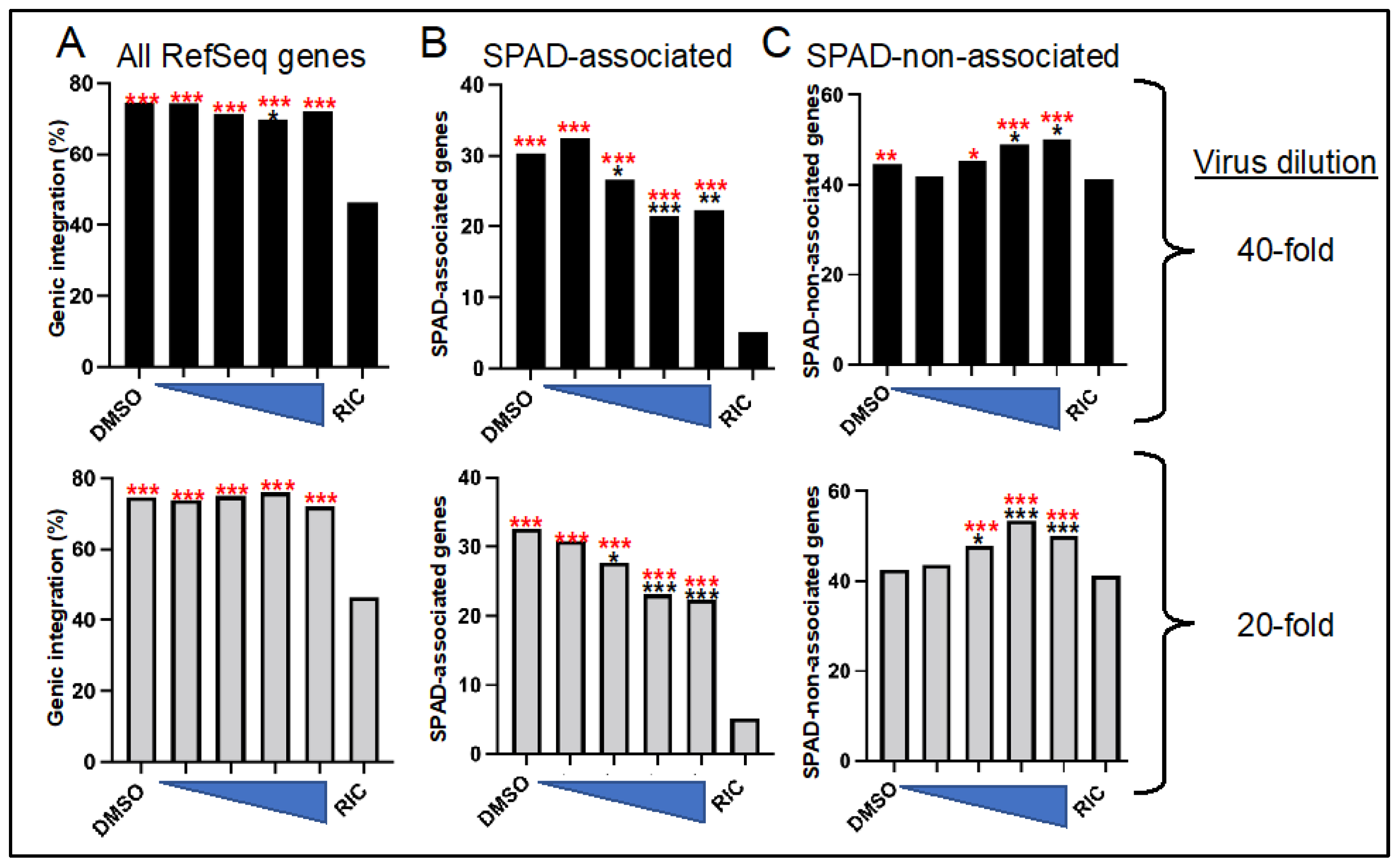
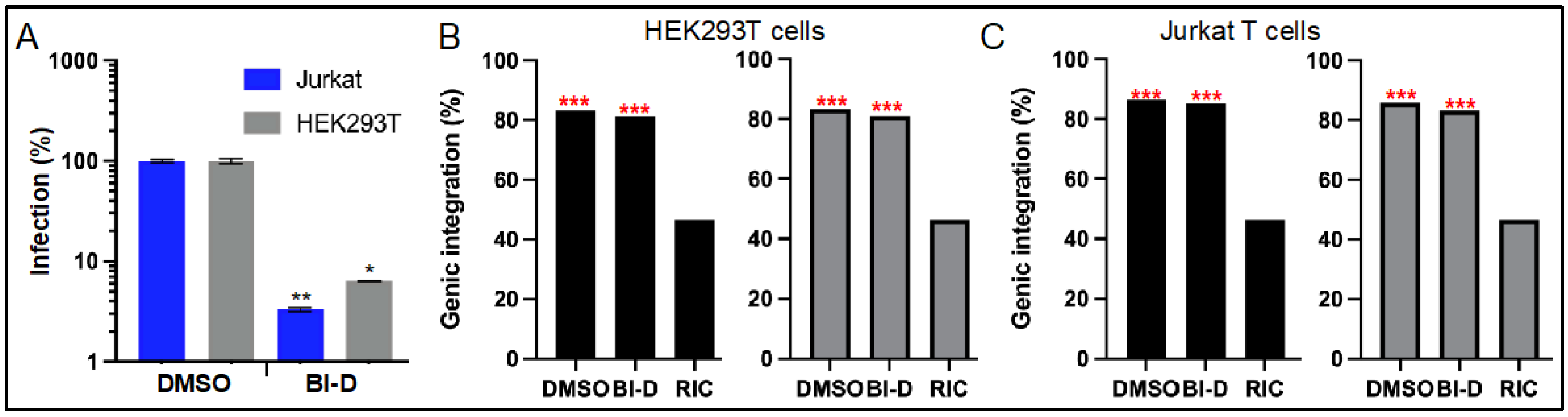
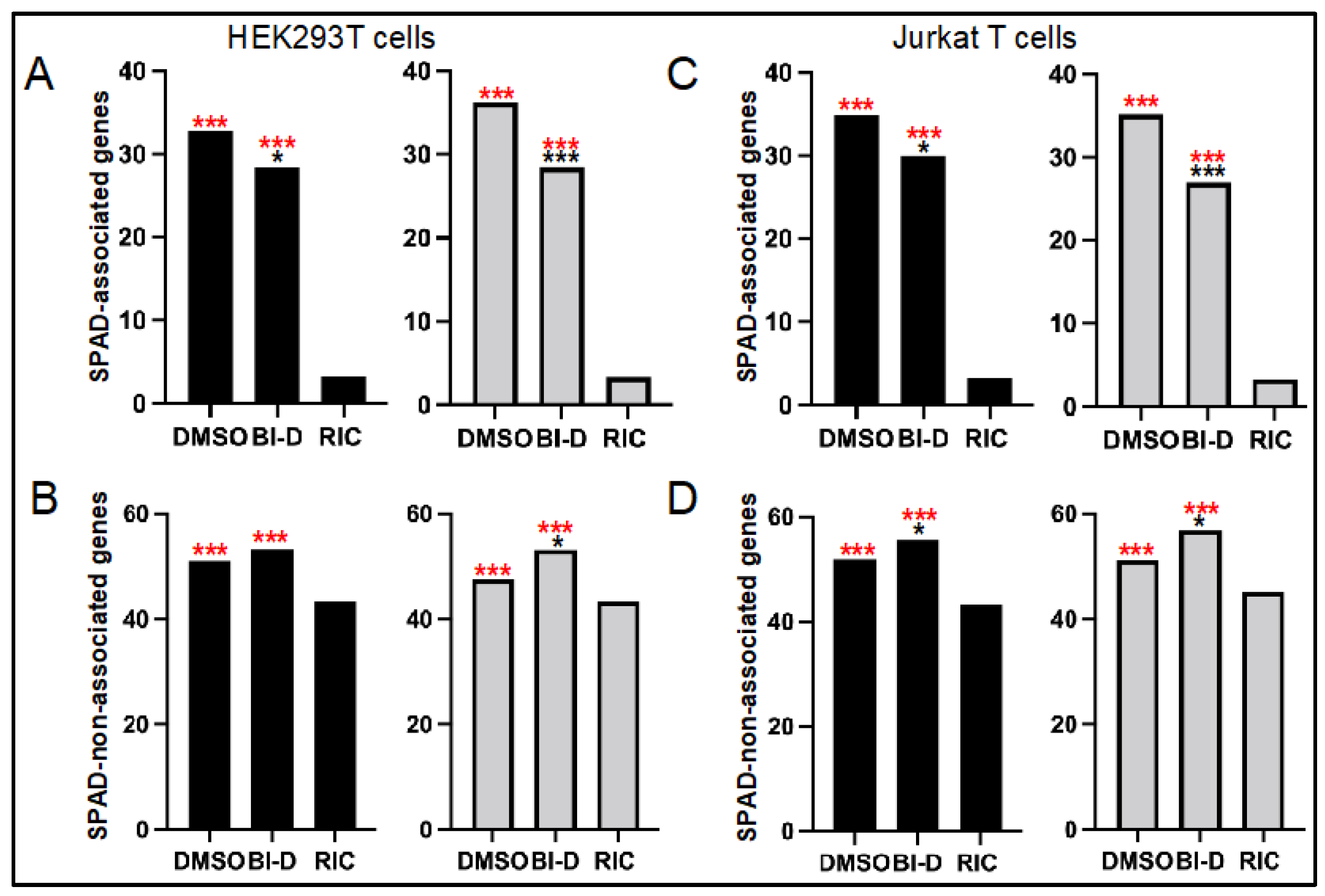
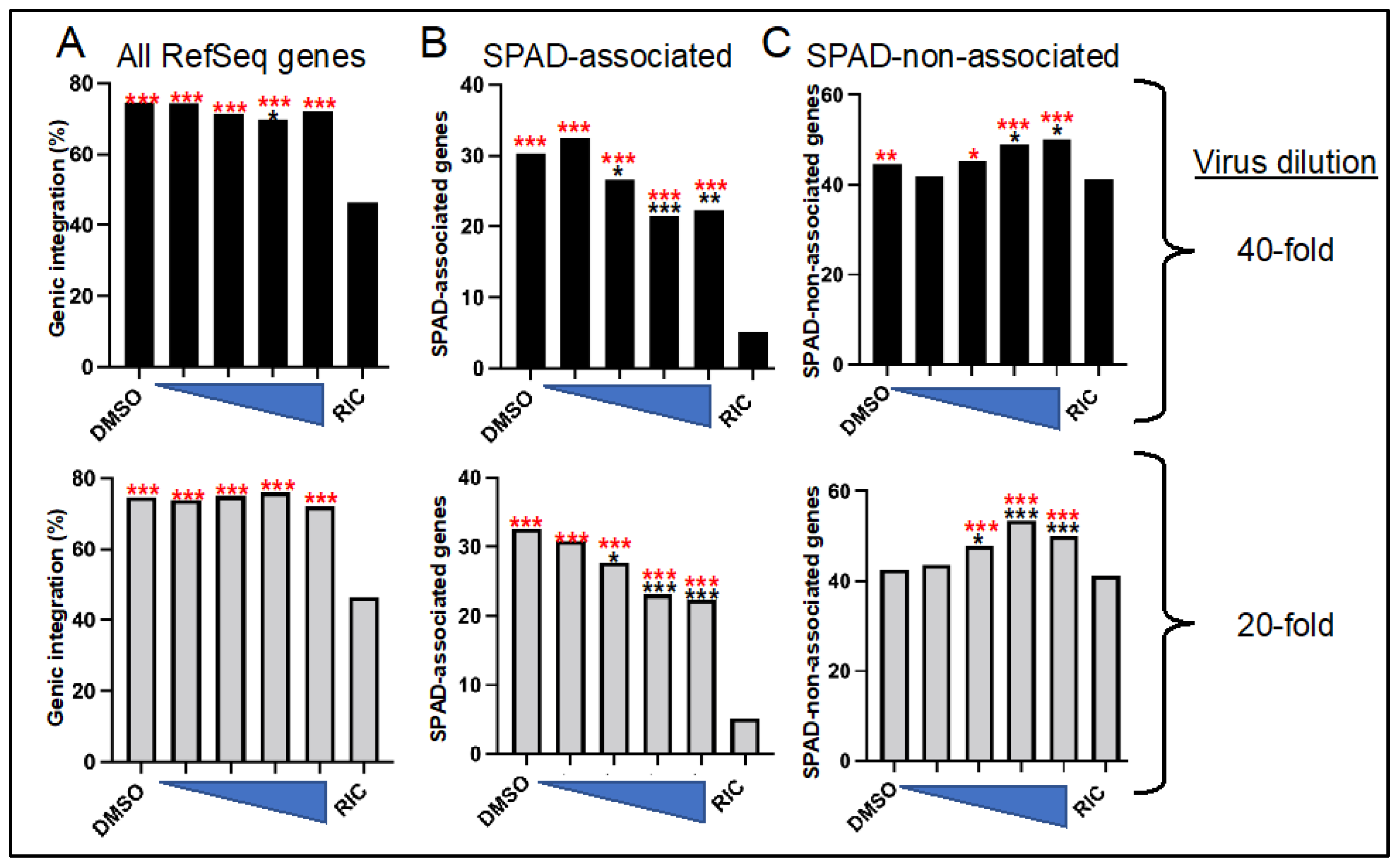
3.3. ALLINI Treated Virions Are Defective for Integration into SPAD-Associated Genes
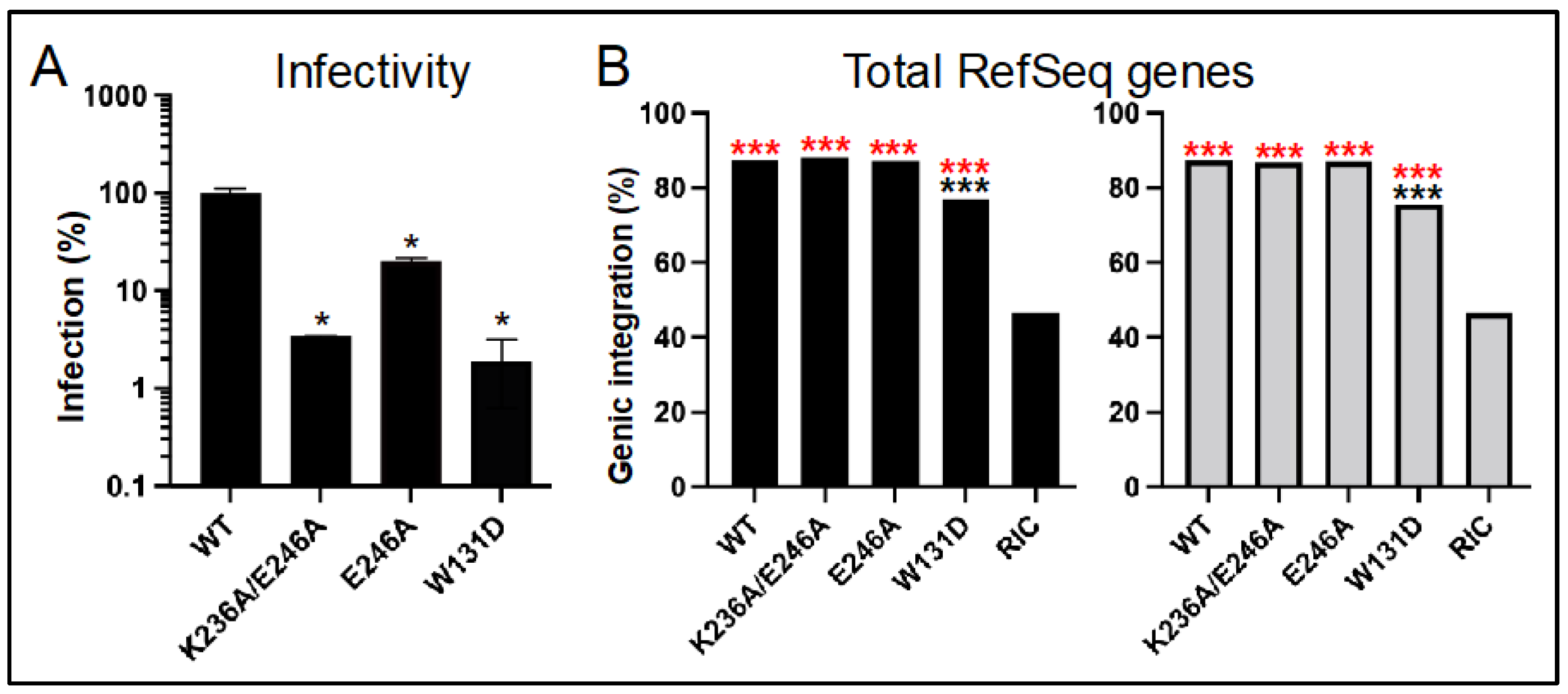
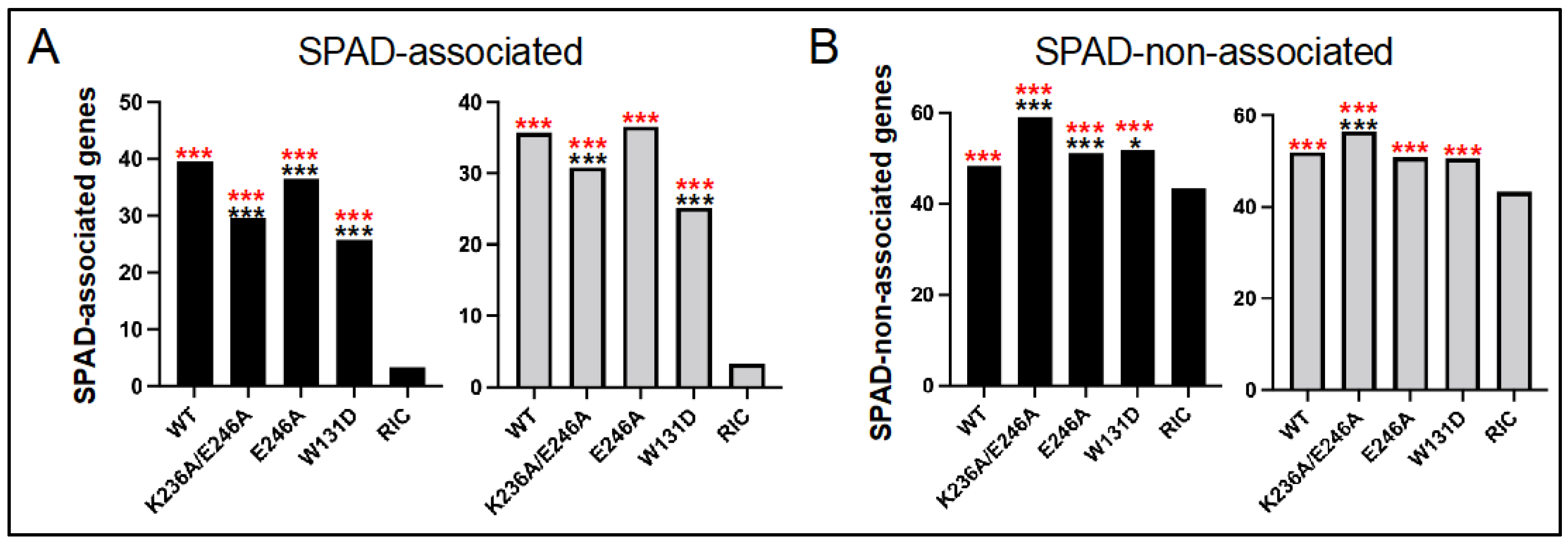
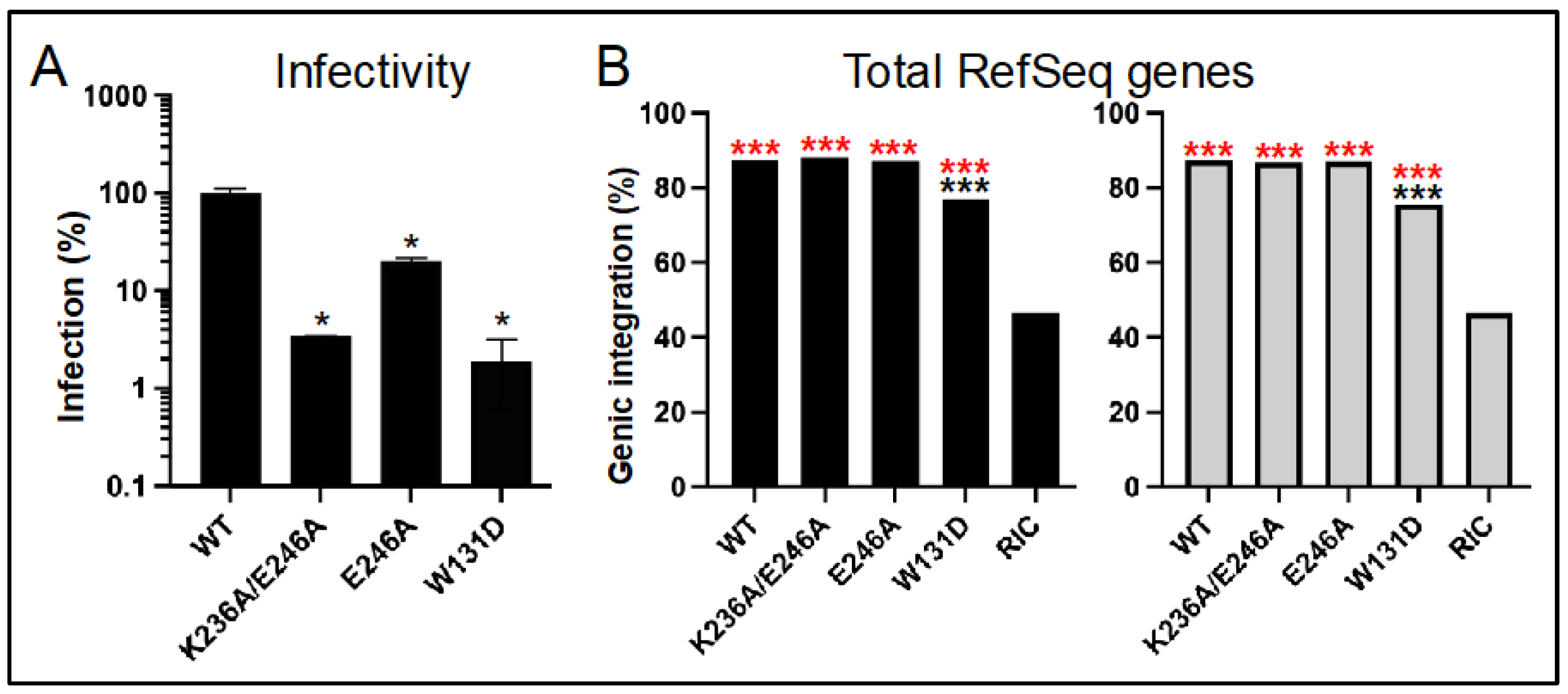
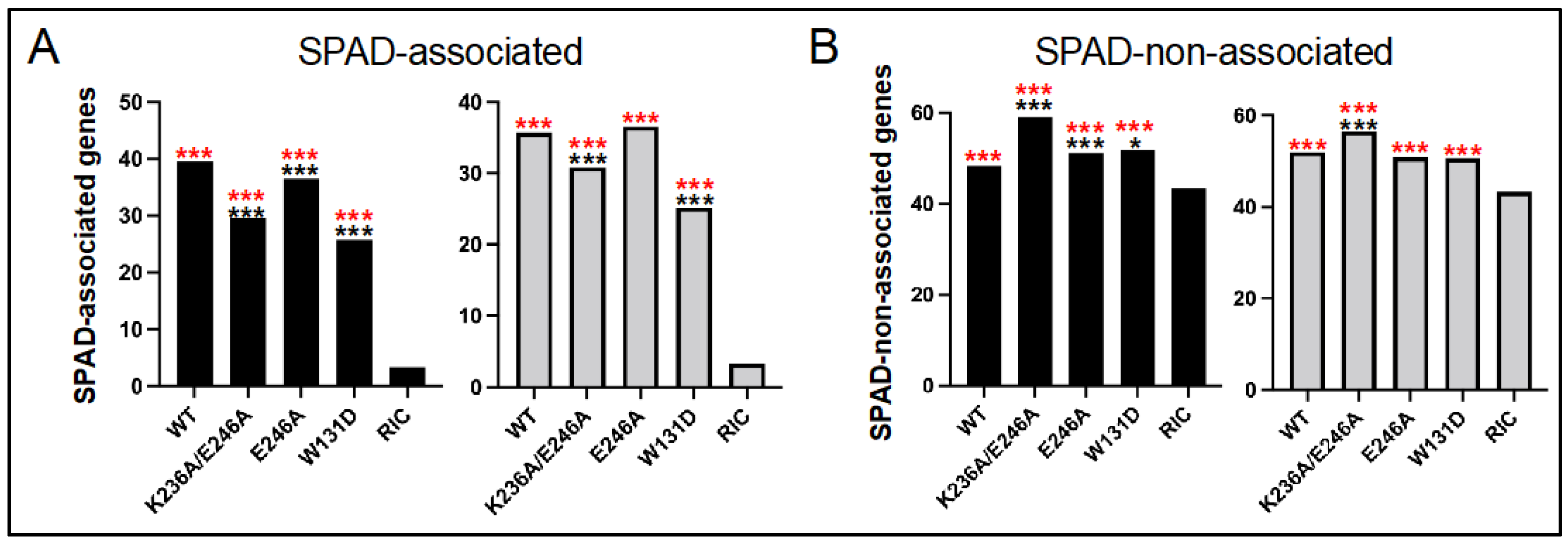
3.4. Class II HIV-1 IN Mutant Viruses Are Defective for Integration into SPAD-Associated Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- De Clercq, E. Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTIs): Past, Present, and Future. Chem. Biodivers. 2004, 1, 44–64. [Google Scholar] [CrossRef] [PubMed]
- Summa, V.; Petrocchi, A.; Bonelli, F.; Crescenzi, B.; Donghi, M.; Ferrara, M.; Fiore, F.; Gardelli, C.; Gonzalez Paz, O.; Hazuda, D.J.; et al. Discovery of Raltegravir, a Potent, Selective Orally Bioavailable HIV-Integrase Inhibitor for the Treatment of HIV-AIDS Infection. J. Med. Chem. 2008, 51, 5843–5855. [Google Scholar] [CrossRef] [PubMed]
- Tsiang, M.; Jones, G.S.; Goldsmith, J.; Mulato, A.; Hansen, D.; Kan, E.; Tsai, L.; Bam, R.A.; Stepan, G.; Stray, K.M.; et al. Antiviral Activity of Bictegravir (GS-9883), a Novel Potent HIV-1 Integrase Strand Transfer Inhibitor with an Improved Resistance Profile. Antimicrob. Agents Chemother. 2016, 60, 7086–7097. [Google Scholar] [CrossRef] [PubMed]
- Hassounah, S.A.; Alikhani, A.; Oliveira, M.; Bharaj, S.; Ibanescu, R.-I.; Osman, N.; Xu, H.-T.; Brenner, B.G.; Mesplède, T.; Wainberg, M.A. Antiviral Activity of Bictegravir and Cabotegravir against Integrase Inhibitor-Resistant SIVmac239 and HIV-1. Antimicrob. Agents Chemother. 2017, 61, e01695-17. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.J.; Zhao, X.Z.; Burke, T.R., Jr.; Hughes, S.H. Efficacies of Cabotegravir and Bictegravir against drug-resistant HIV-1 integrase mutants. Retrovirology 2018, 15, 37. [Google Scholar] [CrossRef]
- Oliveira, M.; Ibanescu, R.I.; Anstett, K.; Mesplede, T.; Routy, J.P.; Robbins, M.A.; Brenner, B.G. Selective resistance profiles emerging in patient-derived clinical isolates with cabotegravir, bictegravir, dolutegravir, and elvitegravir. Retrovirology 2018, 15, 56. [Google Scholar] [CrossRef]
- Saag, M.S.; Gandhi, R.T.; Hoy, J.F.; Landovitz, R.J.; Thompson, M.A.; Sax, P.E.; Smith, D.M.; Benson, C.A.; Buchbinder, S.P.; Del Rio, C.; et al. Antiretroviral drugs for treatment and prevention of HIV infection in adults: 2020 recommendations of the International Antiviral Society-USA Panel. JAMA 2020, 324, 1651–1669. [Google Scholar] [CrossRef]
- Ndashimye, E.; Li, Y.; Reyes, P.S.; Avino, M.; Olabode, A.S.; Kityo, C.M.; Kyeyune, F.; Nankya, I.; Quiñones-Mateu, M.E.; Barr, S.D.; et al. High-level resistance to bictegravir and cabotegravir in subtype A- and D-infected HIV-1 patients failing raltegravir with multiple resistance mutations. J. Antimicrob. Chemother. 2021, 76, 2965–2974. [Google Scholar] [CrossRef]
- Vavro, C.; Ruel, T.; Wiznia, A.; Montañez, N.; Nangle, K.; Horton, J.; Buchanan, A.M.; Stewart, E.L.; Palumbo, P. Emergence of Resistance in HIV-1 Integrase with Dolutegravir Treatment in a Pediatric Population from the IMPAACT P1093 Study. Antimicrob. Agents Chemother. 2022, 66, e0164521. [Google Scholar] [CrossRef]
- Huik, K.; Hill, S.; George, J.; Pau, A.; Kuriakose, S.; Lange, C.M.; Dee, N.; Stoll, P.; Khan, M.; Rehman, T.; et al. High-level dolutegravir resistance can emerge rapidly from few variants and spread by recombination: Implications for INSTI salvage therapy. AIDS, 2022, in press. [CrossRef]
- Hazuda, D.J.; Felock, P.; Witmer, M.; Wolfe, A.; Stillmock, K.; Grobler, J.A.; Espeseth, A.; Gabryelski, L.; Schleif, W.; Blau, C.; et al. Inhibitors of Strand Transfer That Prevent Integration and Inhibit HIV-1 Replication in Cells. Science 2000, 287, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Espeseth, A.S.; Felock, P.; Wolfe, A.; Witmer, M.; Grobler, J.; Anthony, N.; Egbertson, M.; Melamed, J.Y.; Young, S.; Hamill, T.; et al. HIV-1 integrase inhibitors that compete with the target DNA substrate define a unique strand transfer conformation for integrase. Proc. Natl. Acad. Sci. USA 2000, 97, 11244–11249. [Google Scholar] [CrossRef] [PubMed]
- Hare, S.; Gupta, S.S.; Valkov, E.; Engelman, A.; Cherepanov, P. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature 2010, 464, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Passos, D.O.; Li, M.; Jóźwik, I.K.; Zhao, X.Z.; Santos-Martins, D.; Yang, R.; Smith, S.J.; Jeon, Y.; Forli, S.; Hughes, S.H.; et al. Structural basis for strand-transfer inhibitor binding to HIV intasomes. Science 2020, 367, 810–814. [Google Scholar] [CrossRef] [PubMed]
- Engelman, A.N.; Maertens, G.N. Virus-host interactions in retrovirus integration. In Retrovirus-Cell Interactions; Parent, L.J., Ed.; Academic Press: San Diego, CA, USA, 2018; pp. 163–198. [Google Scholar] [CrossRef]
- Schröder, A.R.W.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F. HIV-1 Integration in the Human Genome Favors Active Genes and Local Hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, Y.; Wang, Y.; Zhang, L.; Brinkman, E.K.; Adam, S.A.; Goldman, R.; van Steensel, B.; Ma, J.; Belmont, A.S. Mapping 3D genome organization relative to nuclear compartments using TSA-Seq as a cytological ruler. J. Cell Biol. 2018, 217, 4025–4048. [Google Scholar] [CrossRef]
- Francis, A.C.; Marin, M.; Singh, P.K.; Achuthan, V.; Prellberg, M.J.; Palermino-Rowland, K.; Lan, S.; Tedbury, P.R.; Sarafianos, S.G.; Engelman, A.N.; et al. HIV-1 replication complexes accumulate in nuclear speckles and integrate into speckle-associated genomic domains. Nat. Commun. 2020, 11, 3505. [Google Scholar] [CrossRef]
- Li, W.; Singh, P.K.; Sowd, G.A.; Bedwell, G.J.; Jang, S.; Achuthan, V.; Oleru, A.V.; Wong, D.; Fadel, H.J.; Lee, K.; et al. CPSF6-Dependent Targeting of Speckle-Associated Domains Distinguishes Primate from Nonprimate Lentiviral Integration. mBio 2020, 11, e02254-20. [Google Scholar] [CrossRef]
- Bedwell, G.J.; Jang, S.; Li, W.; Singh, P.K.; Engelman, A.N. rigrag: High-resolution mapping of genic targeting preferences during HIV-1 integration in vitro and in vivo. Nucleic Acids Res. 2021, 49, 7330–7346. [Google Scholar] [CrossRef]
- Singh, P.K.; Bedwell, G.J.; Engelman, A.N. Spatial and Genomic Correlates of HIV-1 Integration Site Targeting. Cells 2022, 11, 655. [Google Scholar] [CrossRef]
- Lee, K.; Ambrose, Z.; Martin, T.D.; Oztop, I.; Mulky, A.; Julias, J.G.; Vandegraaff, N.; Baumann, J.G.; Wang, R.; Yuen, W.; et al. Flexible Use of Nuclear Import Pathways by HIV-1. Cell Host Microbe 2010, 7, 221–233. [Google Scholar] [CrossRef]
- Chin, C.R.; Perreira, J.M.; Savidis, G.; Portmann, J.M.; Aker, A.M.; Feeley, E.M.; Smith, M.C.; Brass, A.L. Direct Visualization of HIV-1 Replication Intermediates Shows that Capsid and CPSF6 Modulate HIV-1 Intra-nuclear Invasion and Integration. Cell Rep. 2015, 13, 1717–1731. [Google Scholar] [CrossRef] [PubMed]
- Achuthan, V.; Perreira, J.M.; Sowd, G.A.; Puray-Chavez, M.; McDougall, W.M.; Paulucci-Holthauzen, A.; Wu, X.; Fadel, H.J.; Poeschla, E.M.; Multani, A.S.; et al. Capsid-CPSF6 Interaction Licenses Nuclear HIV-1 Trafficking to Sites of Viral DNA Integration. Cell Host Microbe 2018, 24, 392–404. [Google Scholar] [CrossRef]
- Cherepanov, P.; Maertens, G.; Proost, P.; Devreese, B.; Van Beeumen, J.; Engelborghs, Y.; De Clercq, E.; Debyser, Z. HIV-1 Integrase Forms Stable Tetramers and Associates with LEDGF/p75 Protein in Human Cells. J. Biol. Chem. 2003, 278, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Llano, M.; Vanegas, M.; Fregoso, O.; Saenz, D.; Chung, S.; Peretz, M.; Poeschla, E.M. LEDGF/p75 Determines Cellular Trafficking of Diverse Lentiviral but Not Murine Oncoretroviral Integrase Proteins and Is a Component of Functional Lentiviral Preintegration Complexes. J. Virol. 2004, 78, 9524–9537. [Google Scholar] [CrossRef] [PubMed]
- Busschots, K.; Vercammen, J.; Emiliani, S.; Benarous, R.; Engelborghs, Y.; Christ, F.; Debyser, Z. The Interaction of LEDGF/p75 with Integrase Is Lentivirus-specific and Promotes DNA Binding. J. Biol. Chem. 2005, 280, 17841–17847. [Google Scholar] [CrossRef]
- Cherepanov, P. LEDGF/p75 interacts with divergent lentiviral integrases and modulates their enzymatic activity in vitro. Nucleic Acids Res. 2007, 35, 113–124. [Google Scholar] [CrossRef]
- Ciuffi, A.; Llano, M.; Poeschla, E.; Hoffmann, C.; Leipzig, J.; Shinn, P.; Ecker, J.R.; Bushman, F. A role for LEDGF/p75 in targeting HIV DNA integration. Nat. Med. 2005, 11, 1287–1289. [Google Scholar] [CrossRef]
- Marshall, H.M.; Ronen, K.; Berry, C.; Llano, M.; Sutherland, H.; Saenz, D.; Bickmore, W.; Poeschla, E.; Bushman, F.D. Role of PSIP1/LEDGF/p75 in Lentiviral Infectivity and Integration Targeting. PLoS ONE 2007, 2, e1340. [Google Scholar] [CrossRef] [Green Version]
- Shun, M.-C.; Raghavendra, N.K.; Vandegraaff, N.; Daigle, J.E.; Hughes, S.; Kellam, P.; Cherepanov, P.; Engelman, A. LEDGF/p75 functions downstream from preintegration complex formation to effect gene-specific HIV-1 integration. Genes Dev. 2007, 21, 1767–1778. [Google Scholar] [CrossRef]
- Singh, P.K.; Plumb, M.R.; Ferris, A.L.; Iben, J.R.; Wu, X.; Fadel, H.J.; Luke, B.T.; Esnault, C.; Poeschla, E.M.; Hughes, S.H.; et al. LEDGF/p75 interacts with mRNA splicing factors and targets HIV-1 integration to highly spliced genes. Genes Dev. 2015, 29, 2287–2297. [Google Scholar] [CrossRef] [PubMed]
- Sowd, G.A.; Serrao, E.; Wang, H.; Wang, W.; Fadel, H.J.; Poeschla, E.M.; Engelman, A.N. A critical role for alternative polyadenylation factor CPSF6 in targeting HIV-1 integration to transcriptionally active chromatin. Proc. Natl. Acad. Sci. USA 2016, 113, E1054–E1063. [Google Scholar] [CrossRef] [PubMed]
- Engelman, A.N.; Kvaratskhelia, M. Multimodal Functionalities of HIV-1 Integrase. Viruses 2022, 14, 926. [Google Scholar] [CrossRef] [PubMed]
- Maertens, G.; Cherepanov, P.; Pluymers, W.; Busschots, K.; De Clercq, E.; Debyser, Z.; Engelborghs, Y. LEDGF/p75 Is Essential for Nuclear and Chromosomal Targeting of HIV-1 Integrase in Human Cells. J. Biol. Chem. 2003, 278, 33528–33539. [Google Scholar] [CrossRef]
- Cherepanov, P.; Ambrosio, A.L.B.; Rahman, S.; Ellenberger, T.; Engelman, A. From the Cover: Structural basis for the recognition between HIV-1 integrase and transcriptional coactivator p75. Proc. Natl. Acad. Sci. USA 2005, 102, 17308–17313. [Google Scholar] [CrossRef]
- Busschots, K.; Voet, A.; De Maeyer, M.; Rain, J.-C.; Emiliani, S.; Benarous, R.; Desender, L.; Debyser, Z.; Christ, F. Identification of the LEDGF/p75 Binding Site in HIV-1 Integrase. J. Mol. Biol. 2007, 365, 1480–1492. [Google Scholar] [CrossRef]
- Rahman, S.; Lu, R.; Vandegraaff, N.; Cherepanov, P.; Engelman, A. Structure-based mutagenesis of the integrase-LEDGF/p75 interface uncouples a strict correlation between in vitro protein binding and HIV-1 fitness. Virology 2007, 357, 79–90. [Google Scholar] [CrossRef]
- Hare, S.; Shun, M.-C.; Gupta, S.S.; Valkov, E.; Engelman, A.; Cherepanov, P. A Novel Co-Crystal Structure Affords the Design of Gain-of-Function Lentiviral Integrase Mutants in the Presence of Modified PSIP1/LEDGF/p75. PLoS Pathog. 2009, 5, e1000259. [Google Scholar] [CrossRef]
- Cherepanov, P.; Devroe, E.; Silver, P.A.; Engelman, A. Identification of an evolutionarily-conserved domain in LEDGF/p75 that binds HIV-1 integrase. J. Biol. Chem. 2004, 279, 48883–48892. [Google Scholar] [CrossRef] [Green Version]
- Vanegas, M.; Llano, M.; Delgado, S.; Thompson, D.; Peretz, M.; Poeschla, E. Identification of the LEDGF/p75 HIV-1 integrase-interaction domain and NLS reveals NLS-independent chromatin tethering. J. Cell Sci. 2005, 118 Pt 8, 1733–1743. [Google Scholar] [CrossRef]
- Izumoto, Y.; Kuroda, T.; Harada, H.; Kishimoto, T.; Nakamura, H. Hepatoma-Derived Growth Factor Belongs to a Gene Family in Mice Showing Significant Homology in the Amino Terminus. Biochem. Biophys. Res. Commun. 1997, 238, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Schrijvers, R.; Vets, S.; De Rijck, J.; Malani, N.; Bushman, F.D.; Debyser, Z.; Gijsbers, R. HRP-2 determines HIV-1 integration site selection in LEDGF/p75 depleted cells. Retrovirology 2012, 9, 84. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Jurado, K.A.; Wu, X.; Shun, M.-C.; Li, X.; Ferris, A.L.; Smith, S.J.; Patel, P.A.; Fuchs, J.R.; Cherepanov, P.; et al. HRP2 determines the efficiency and specificity of HIV-1 integration in LEDGF/p75 knockout cells but does not contribute to the antiviral activity of a potent LEDGF/p75-binding site integrase inhibitor. Nucleic Acids Res. 2012, 40, 11518–11530. [Google Scholar] [CrossRef] [PubMed]
- Madison, M.K.; Lawson, D.Q.; Elliott, J.; Ozantürk, A.N.; Koneru, P.C.; Townsend, D.; Errando, M.; Kvaratskhelia, M.; Kutluay, S.B. Allosteric HIV-1 Integrase Inhibitors Lead to Premature Degradation of the Viral RNA Genome and Integrase in Target Cells. J. Virol. 2017, 91, e00821-17. [Google Scholar] [CrossRef] [PubMed]
- Koneru, P.C.; Francis, A.C.; Deng, N.; Rebensburg, S.V.; Hoyte, A.C.; Lindenberger, J.; Adu-Ampratwum, D.; Larue, R.C.; Wempe, M.F.; Engelman, A.N.; et al. HIV-1 integrase tetramers are the antiviral target of pyridine-based allosteric integrase inhibitors. eLife 2019, 8, e46344. [Google Scholar] [CrossRef]
- Elliott, J.L.; Eschbach, J.E.; Koneru, P.C.; Li, W.; Puray-Chavez, M.; Townsend, D.; Lawson, D.Q.; Engelman, A.N.; Kvaratskhelia, M.; Kutluay, S.B. Integrase-RNA interactions underscore the critical role of integrase in HIV-1 virion morphogenesis. elife 2020, 9, e54311. [Google Scholar] [CrossRef]
- Engelman, A. In Vivo Analysis of Retroviral Integrase Structure and Function. Adv. Virus Res. 1999, 52, 411–426. [Google Scholar] [CrossRef]
- Kessl, J.J.; Kutluay, S.B.; Townsend, D.; Rebensburg, S.; Slaughter, A.; Larue, R.C.; Shkriabai, N.; Bakouche, N.; Fuchs, J.R.; Bieniasz, P.D.; et al. HIV-1 Integrase Binds the Viral RNA Genome and Is Essential during Virion Morphogenesis. Cell 2016, 166, 1257–1268. [Google Scholar] [CrossRef]
- Kessl, J.J.; Jena, N.; Koh, Y.; Taskent-Sezgin, H.; Slaughter, A.; Feng, L.; de Silva, S.; Wu, L.; Le Grice, S.F.J.; Engelman, A.; et al. Multimode, Cooperative Mechanism of Action of Allosteric HIV-1 Integrase Inhibitors. J. Biol. Chem. 2012, 287, 16801–16811. [Google Scholar] [CrossRef] [Green Version]
- Christ, F.; Voet, A.; Marchand, A.; Nicolet, S.; Desimmie, B.A.; Marchand, D.; Bardiot, D.; Van Der Veken, N.J.; Van Remoortel, B.; Strelkov, S.V.; et al. Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication. Nat. Chem. Biol. 2010, 6, 442–448. [Google Scholar] [CrossRef]
- Balakrishnan, M.; Yant, S.R.; Tsai, L.; O’Sullivan, C.; Bam, R.A.; Tsai, A.; Niedziela-Majka, A.; Stray, K.M.; Sakowicz, R.; Cihlar, T. Non-Catalytic Site HIV-1 Integrase Inhibitors Disrupt Core Maturation and Induce a Reverse Transcription Block in Target Cells. PLoS ONE 2013, 8, e74163. [Google Scholar] [CrossRef] [PubMed]
- Le Rouzic, E.; Bonnard, D.; Chasset, S.; Bruneau, J.-M.; Chevreuil, F.; Le Strat, F.; Nguyen, J.; Beauvoir, R.; Amadori, C.; Brias, J.; et al. Dual inhibition of HIV-1 replication by integrase-LEDGF allosteric inhibitors is predominant at the post-integration stage. Retrovirology 2013, 10, 144. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Slaughter, A.; Jena, N.; Feng, L.; Kessl, J.J.; Fadel, H.J.; Malani, N.; Male, F.; Wu, L.; Poeschla, E.; et al. A New Class of Multimerization Selective Inhibitors of HIV-1 Integrase. PLoS Pathog. 2014, 10, e1004171. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Dharmarajan, V.; Serrao, E.; Hoyte, A.; LaRue, R.C.; Slaughter, A.; Sharma, A.; Plumb, M.R.; Kessl, J.J.; Fuchs, J.R.; et al. The Competitive Interplay between Allosteric HIV-1 Integrase Inhibitor BI/D and LEDGF/p75 during the Early Stage of HIV-1 Replication Adversely Affects Inhibitor Potency. ACS Chem. Biol. 2016, 11, 1313–1321. [Google Scholar] [CrossRef]
- Vranckx, L.S.; Demeulemeester, J.; Saleh, S.; Boll, A.; Vansant, G.; Schrijvers, R.; Weydert, C.; Battivelli, E.; Verdin, E.; Cereseto, A.; et al. LEDGIN-mediated Inhibition of Integrase–LEDGF/p75 Interaction Reduces Reactivation of Residual Latent HIV. EBioMedicine 2016, 8, 248–264. [Google Scholar] [CrossRef]
- Feng, L.; Sharma, A.; Slaughter, A.; Jena, N.; Koh, Y.; Shkriabai, N.; Larue, R.C.; Patel, P.A.; Mitsuya, H.; Kessl, J.J.; et al. The A128T Resistance Mutation Reveals Aberrant Protein Multimerization as the Primary Mechanism of Action of Allosteric HIV-1 Integrase Inhibitors. J. Biol. Chem. 2013, 288, 15813–15820. [Google Scholar] [CrossRef]
- Deng, N.; Hoyte, A.; Mansour, Y.E.; Mohamed, M.S.; Fuchs, J.R.; Engelman, A.N.; Kvaratskhelia, M.; Levy, R. Allosteric HIV-1 integrase inhibitors promote aberrant protein multimerization by directly mediating inter-subunit interactions: Structural and thermodynamic modeling studies. Protein Sci. 2016, 25, 1911–1917. [Google Scholar] [CrossRef]
- Gupta, K.; Turkki, V.; Sherrill-Mix, S.; Hwang, Y.; Eilers, G.; Taylor, L.; McDanal, C.; Wang, P.; Temelkoff, D.; Nolte, R.T.; et al. Structural Basis for Inhibitor-Induced Aggregation of HIV Integrase. PLoS Biol. 2016, 14, e1002584. [Google Scholar] [CrossRef]
- Gupta, K.; Allen, A.; Giraldo, C.; Eilers, G.; Sharp, R.; Hwang, Y.; Murali, H.; Cruz, K.; Janmey, P.; Bushman, F.; et al. Allosteric HIV Integrase Inhibitors Promote Formation of Inactive Branched Polymers via Homomeric Carboxy-Terminal Domain Interactions. Structure 2021, 29, 213–225. [Google Scholar] [CrossRef]
- Jurado, K.A.; Wang, H.; Slaughter, A.; Feng, L.; Kessl, J.J.; Koh, Y.; Wang, W.; Ballandras-Colas, A.; Patel, P.A.; Fuchs, J.R.; et al. Allosteric integrase inhibitor potency is determined through the inhibition of HIV-1 particle maturation. Proc. Natl. Acad. Sci. USA 2013, 110, 8690–8695. [Google Scholar] [CrossRef]
- Fadel, H.J.; Morrison, J.H.; Saenz, D.T.; Fuchs, J.R.; Kvaratskhelia, M.; Ekker, S.C.; Poeschla, E.M. TALEN Knockout of the PSIP1 Gene in Human Cells: Analyses of HIV-1 Replication and Allosteric Integrase Inhibitor Mechanism. J. Virol. 2014, 88, 9704–9717. [Google Scholar] [CrossRef] [PubMed]
- Ballandras-Colas, A.; Chivukula, V.; Gruszka, D.T.; Shan, Z.; Singh, P.K.; Pye, V.E.; McLean, R.K.; Bedwell, G.J.; Li, W.; Nans, A.; et al. Multivalent interactions essential for lentiviral integrase function. Nat. Commun. 2022, 13, 2416. [Google Scholar] [CrossRef] [PubMed]
- VanSant, G.; Vranckx, L.S.; Zurnic, I.; Van Looveren, D.; Van De Velde, P.; Nobles, C.; Gijsbers, R.; Christ, F.; Debyser, Z. Impact of LEDGIN treatment during virus production on residual HIV-1 transcription. Retrovirology 2019, 16, 8. [Google Scholar] [CrossRef]
- Bruggemans, A.; Vansant, G.; Balakrishnan, M.; Mitchell, M.L.; Cai, R.; Christ, F.; Debyser, Z. GS-9822, a Preclinical LEDGIN Candidate, Displays a Block-and-Lock Phenotype in Cell Culture. Antimicrob. Agents Chemother. 2021, 65, e02328-20. [Google Scholar] [CrossRef]
- Maehigashi, T.; Ahn, S.; Kim, U.-I.; Lindenberger, J.; Oo, A.; Koneru, P.C.; Mahboubi, B.; Engelman, A.N.; Kvaratskhelia, M.; Kim, K.; et al. A highly potent and safe pyrrolopyridine-based allosteric HIV-1 integrase inhibitor targeting host LEDGF/p75-integrase interaction site. PLoS Pathog. 2021, 17, e1009671. [Google Scholar] [CrossRef]
- Naidu, B.N.; Patel, M.; McAuliffe, B.; Ding, B.; Cianci, C.; Simmermacher, J.; Jenkins, S.; Parker, D.D.; Sivaprakasam, P.; Khan, J.A.; et al. Design, synthesis, and preclinical profiling of GSK3739936 (BMS-986180), an allosteric inhibitor of HIV-1 integrase with broad-spectrum activity toward 124/125 polymorphs. J. Med. Chem. 2022, 65, 4949–4971. [Google Scholar] [CrossRef]
- Ohata, Y.; Tomonaga, M.; Watanabe, Y.; Tomura, K.; Kimura, K.; Akaki, T.; Adachi, K.; Kodama, E.N.; Matsuzaki, Y.; Hayashi, H. Antiviral Activity and Resistance Profile of the Novel HIV-1 Non-Catalytic Site Integrase Inhibitor JTP-0157602. J. Virol. 2022, 96, e0184321. [Google Scholar] [CrossRef] [PubMed]
- Parcella, K.; Wang, T.; Eastman, K.; Zhang, Z.; Yin, Z.; Patel, M.; Tu, Y.; Zheng, B.Z.; Walker, M.A.; Saulnier, M.G.; et al. Discovery and Preclinical Profiling of GSK3839919, a Potent HIV-1 Allosteric Integrase Inhibitor. ACS Med. Chem. Lett. 2022, 13, 972–980. [Google Scholar] [CrossRef]
- Parcella, K.; Patel, M.; Tu, Y.; Eastman, K.; Peese, K.; Gillis, E.; Belema, M.; Dicker, I.B.; McAuliffe, B.; Ding, B.; et al. Scaffold modifications to the 4-(4,4-dimethylpiperidinyl) 2,6-dimethylpyridinyl class of HIV-1 allosteric integrase inhibitors. Bioorg. Med. Chem. 2022, 67, 116833. [Google Scholar] [CrossRef]
- Lu, R.; Limón, A.; Devroe, E.; Silver, P.A.; Cherepanov, P.; Engelman, A. Class II Integrase Mutants with Changes in Putative Nuclear Localization Signals Are Primarily Blocked at a Postnuclear Entry Step of Human Immunodeficiency Virus Type 1 Replication. J. Virol. 2004, 78, 12735–12746. [Google Scholar] [CrossRef]
- Koh, Y.; Wu, X.; Ferris, A.L.; Matreyek, K.A.; Smith, S.J.; Lee, K.; KewalRamani, V.N.; Hughes, S.H.; Engelman, A. Differential Effects of Human Immunodeficiency Virus Type 1 Capsid and Cellular Factors Nucleoporin 153 and LEDGF/p75 on the Efficiency and Specificity of Viral DNA Integration. J. Virol. 2013, 87, 648–658. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Ghory, H.Z.; Engelman, A. Genetic Analyses of Conserved Residues in the Carboxyl-Terminal Domain of Human Immunodeficiency Virus Type 1 Integrase. J. Virol. 2005, 79, 10356–10368. [Google Scholar] [CrossRef] [PubMed]
- Shun, M.-C.; Daigle, J.E.; Vandegraaff, N.; Engelman, A. Wild-Type Levels of Human Immunodeficiency Virus Type 1 Infectivity in the Absence of Cellular Emerin Protein. J. Virol. 2007, 81, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Cook, N.J.; Li, W.; Berta, D.; Badaoui, M.; Ballandras-Colas, A.; Nans, A.; Kotecha, A.; Rosta, E.; Engelman, A.N.; Cherepanov, P. Structural basis of second-generation HIV integrase inhibitor action and viral resistance. Science 2020, 367, 806–810. [Google Scholar] [CrossRef]
- Matreyek, K.A.; Wang, W.; Serrao, E.; Singh, P.K.; Levin, H.L.; Engelman, A. Host and viral determinants for MxB restriction of HIV-1 infection. Retrovirology 2014, 11, 90. [Google Scholar] [CrossRef]
- Serrao, E.; Cherepanov, P.; Engelman, A.N. Amplification, Next-generation Sequencing, and Genomic DNA Mapping of Retroviral Integration Sites. J. Vis. Exp. 2016, 109, e53840. [Google Scholar] [CrossRef]
- Anderson-Daniels, J.; Singh, P.K.; Sowd, G.A.; Li, W.; Engelman, A.N.; Aiken, C. Dominant Negative MA-CA Fusion Protein Is Incorporated into HIV-1 Cores and Inhibits Nuclear Entry of Viral Preintegration Complexes. J. Virol. 2019, 93, e01118-19. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows—Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Tsiang, M.; Jones, G.S.; Niedziela-Majka, A.; Kan, E.; Lansdon, E.B.; Huang, W.; Hung, M.; Samuel, D.; Novikov, N.; Xu, Y.; et al. New Class of HIV-1 Integrase (IN) Inhibitors with a Dual Mode of Action. J. Biol. Chem. 2012, 287, 21189–21203. [Google Scholar] [CrossRef] [PubMed]
- Schrijvers, R.; De Rijck, J.; Demeulemeester, J.; Adachi, N.; Vets, S.; Ronen, K.; Christ, F.; Bushman, F.D.; Debyser, Z.; Gijsbers, R. LEDGF/p75-independent HIV-1 replication demonstrates a role for HRP-2 and remains sensitive to inhibition by LEDGINs. PLoS Pathog. 2012, 8, e1002558. [Google Scholar] [CrossRef] [PubMed]
- Fader, L.D.; Malenfant, E.; Parisien, M.; Carson, R.; Bilodeau, F.; Landry, S.; Pesant, M.; Brochu, C.; Morin, S.; Chabot, C.; et al. Discovery of BI 224436, a Noncatalytic Site Integrase Inhibitor (NCINI) of HIV-1. ACS Med. Chem. Lett. 2014, 5, 422–427. [Google Scholar] [CrossRef]
- Gupta, K.; Brady, T.; Dyer, B.M.; Malani, N.; Hwang, Y.; Male, F.; Nolte, R.T.; Wang, L.; Velthuisen, E.; Jeffrey, J.; et al. Allosteric inhibition of human immunodeficiency virus integrase: Late block during viral replication and abnormal multimerization involving specific protein domains. J. Biol. Chem. 2014, 289, 20477–20488. [Google Scholar] [CrossRef]
- Desimmie, B.A.; Schrijvers, R.; Demeulemeester, J.; Borrenberghs, D.; Weydert, C.; Thys, W.; Vets, S.; Van Remoortel, B.; Hofkens, J.; De Rijck, J.; et al. LEDGINs inhibit late stage HIV-1 replication by modulating integrase multimerization in the virions. Retrovirology 2013, 10, 57. [Google Scholar] [CrossRef]
- Fontana, J.; Jurado, K.A.; Cheng, N.; Ly, N.L.; Fuchs, J.R.; Gorelick, R.J.; Engelman, A.N.; Steven, A.C. Distribution and Redistribution of HIV-1 Nucleocapsid Protein in Immature, Mature, and Integrase-Inhibited Virions: A Role for Integrase in Maturation. J. Virol. 2015, 89, 9765–9780. [Google Scholar] [CrossRef]
- Christ, F.; Shaw, S.; Demeulemeester, J.; Desimmie, B.A.; Marchand, A.; Butler, S.; Smets, W.; Chaltin, P.; Westby, M.; Debyser, Z.; et al. Small molecule inhibitors of the LEDGF/p75 binding site of integrase (LEDGINs) block HIV replication and modulate integrase multimerization. Antimicrob. Agents Chemother. 2012, 56, 4365–4374. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Limón, A.; Ghory, H.Z.; Engelman, A. Genetic Analyses of DNA-Binding Mutants in the Catalytic Core Domain of Human Immunodeficiency Virus Type 1 Integrase. J. Virol. 2005, 79, 2493–2505. [Google Scholar] [CrossRef]
- Mohammed, K.D.; Topper, M.B.; Muesing, M.A. Sequential Deletion of the Integrase (Gag-Pol) Carboxyl Terminus Reveals Distinct Phenotypic Classes of Defective HIV-1. J. Virol. 2011, 85, 4654–4666. [Google Scholar] [CrossRef] [Green Version]
- Debyser, Z.; Bruggemans, A.; Van Belle, S.; Janssens, J.; Christ, F. LEDGINs, Inhibitors of the Interaction between HIV-1 Integrase and LEDGF/p75, Are Potent Antivirals with a Potential to Cure HIV Infection. Adv. Exp. Med. Biol. 2021, 1322, 97–114. [Google Scholar] [CrossRef]
- McKee, C.J.; Kessl, J.J.; Shkriabai, N.; Dar, M.J.; Engelman, A.; Kvaratskhelia, M. Dynamic Modulation of HIV-1 Integrase Structure and Function by Cellular Lens Epithelium-derived Growth Factor (LEDGF) Protein. J. Biol. Chem. 2008, 283, 31802–31812. [Google Scholar] [CrossRef]
- Burdick, R.C.; Li, C.; Munshi, M.; Rawson, J.M.O.; Nagashima, K.; Hu, W.-S.; Pathak, V.K. HIV-1 uncoats in the nucleus near sites of integration. Proc. Natl. Acad. Sci. USA 2020, 117, 5486–5493. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Burdick, R.C.; Nagashima, K.; Hu, W.-S.; Pathak, V.K. HIV-1 cores retain their integrity until minutes before uncoating in the nucleus. Proc. Natl. Acad. Sci. USA 2021, 118, e2019467118. [Google Scholar] [CrossRef] [PubMed]
- Achuthan, V.; Perreira, J.M.; Ahn, J.J.; Brass, A.L.; Engelman, A.N. Capsid-CPSF6 interaction: Master regulator of nuclear HIV-1 positioning and integration. J. Life Sci. (Westlake Village) 2019, 1, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Cereseto, A.; Manganaro, L.; Gutierrez, M.I.; Terreni, M.; Fittipaldi, A.; Lusic, M.; Marcello, A.; Giacca, M. Acetylation of HIV-1 integrase by p300 regulates viral integration. EMBO J. 2005, 24, 3070–3081. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | BI-D | Replicate 1 | Replicate 2 | ||
|---|---|---|---|---|---|
| Number | In genes (%) | Number | In genes (%) | ||
| WT | – 2 | 1062 | 82.0 | 5100 | 80.4 |
| EC70 | 1971 | 69.1 | 4465 | 66.4 | |
| EC95 | 2131 | 69.1 | 1969 | 70.4 | |
| HKO | – | 16,266 | 81.7 | 10,767 | 82.4 |
| EC70 | 7299 | 67.3 | 6998 | 67.8 | |
| EC95 | 4869 | 69.9 | 3154 | 68.9 | |
| LKO | – | 3673 | 65.1 | 1755 | 63.9 |
| EC70 | 694 | 65.1 | 355 | 63.9 | |
| EC95 | 246 | 55.7 | 303 | 55.4 | |
| DKO | – | 1936 | 54.2 | 1639 | 55.5 |
| EC70 | 559 | 56.5 | 2170 | 54.7 | |
| EC95 | 485 | 51.3 | 1171 | 53.5 | |
| RIC 3 | n.a. 4 | 112,183 | 46.5 | 112,183 | 46.5 |
| Infected Cell | Producer Cell Treatment | Replicate 1 | Replicate 2 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Number | Refseq Genes | SPADg | SPAD-Nong | Number | Refseq Genes | SPADg | SPAD-Nong | ||
| HEK293T | DMSO | 14,718 | 83.3 | 32.8 | 51.0 | 23,448 | 83.3 | 36.2 | 47.6 |
| BI-D | 676 | 81.4 | 28.4 | 53.3 | 914 | 81.1 | 28.5 | 53.2 | |
| Jurkat | DMSO | 14,583 | 86.3 | 34.9 | 51.8 | 27,044 | 85.9 | 35.2 | 51.2 |
| BI-D | 812 | 85.2 | 29.9 | 55.7 | 654 | 83.3 | 26.9 | 56.9 | |
| RIC 2 | n.a. 3 | 112,183 | 46.5 | 3.3 | 43.3 | 112,183 | 46.5 | 3.3 | 43.3 |
| CX014442 (nM) | 1-to-40 Dilution | 1-to-20 Dilution | ||||||
|---|---|---|---|---|---|---|---|---|
| Number | In Genes (%) | In SPADg (%) | In SPAD-Nong (%) | Number | In genes (%) | In SPADg (%) | In SPAD-Nong (%) | |
| – 2 | 2725 | 74.4 | 30.3 | 44.5 | 3530 | 74.7 | 32.6 | 42.6 |
| 31.25 | 1282 | 74.3 | 32.5 | 41.8 | 1331 | 73.9 | 30.8 | 43.7 |
| 62.5 | 979 | 71.5 | 26.6 | 45.3 | 1028 | 75.0 | 27.7 | 47.9 |
| 125 | 1267 | 69.9 | 21.5 | 48.9 | 576 | 76.0 | 23.1 | 53.5 |
| 250 | 586 | 72.2 | 22.4 | 50.2 | 935 | 72.1 | 22.4 | 50.1 |
| RIC 3 | 9,133,735 | 46.4 | 4.9 | 41.7 | 9,133,735 | 46.4 | 4.9 | 41.7 |
| IN | Replicate 1 | Replicate 2 | ||||||
|---|---|---|---|---|---|---|---|---|
| Number | In Genes (%) | In SPADg (%) | In SPAD-Nong (%) | Number | In Genes (%) | In SPADg (%) | In SPAD-Nong (%) | |
| WT | 7342 | 87.3 | 39.5 | 48.3 | 17,554 | 87.3 | 35.7 | 52.0 |
| K236A/E246A | 4754 | 88.1 | 29.7 | 59.1 | 2216 | 86.9 | 30.9 | 56.5 |
| E246A | 19,827 | 87.1 | 36.4 | 51.1 | 10,899 | 87.0 | 36.6 | 50.8 |
| W131D | 2347 | 77.0 | 25.8 | 51.7 | 3552 | 75.5 | 25.2 | 50.7 |
| RIC 2 | 112,183 | 46.5 | 3.3 | 43.3 | 112,183 | 46.5 | 3.3 | 43.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, P.K.; Li, W.; Bedwell, G.J.; Fadel, H.J.; Poeschla, E.M.; Engelman, A.N. Allosteric Integrase Inhibitor Influences on HIV-1 Integration and Roles of LEDGF/p75 and HDGFL2 Host Factors. Viruses 2022, 14, 1883. https://doi.org/10.3390/v14091883
Singh PK, Li W, Bedwell GJ, Fadel HJ, Poeschla EM, Engelman AN. Allosteric Integrase Inhibitor Influences on HIV-1 Integration and Roles of LEDGF/p75 and HDGFL2 Host Factors. Viruses. 2022; 14(9):1883. https://doi.org/10.3390/v14091883
Chicago/Turabian StyleSingh, Parmit Kumar, Wen Li, Gregory J. Bedwell, Hind J. Fadel, Eric M. Poeschla, and Alan N. Engelman. 2022. "Allosteric Integrase Inhibitor Influences on HIV-1 Integration and Roles of LEDGF/p75 and HDGFL2 Host Factors" Viruses 14, no. 9: 1883. https://doi.org/10.3390/v14091883
APA StyleSingh, P. K., Li, W., Bedwell, G. J., Fadel, H. J., Poeschla, E. M., & Engelman, A. N. (2022). Allosteric Integrase Inhibitor Influences on HIV-1 Integration and Roles of LEDGF/p75 and HDGFL2 Host Factors. Viruses, 14(9), 1883. https://doi.org/10.3390/v14091883