
Genomic Surveillance of SARS-CoV-2 in the Southern Province of Zambia: Detection and Characterization of Alpha, Beta, Delta, and Omicron Variants of Concern

 , , , , , , ,
, , , , , , ,  ,
,  , , and
, , and  add
Show full author list
add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Site and Sample Collection
2.2. RNA Extraction and Virus Genome Amplification
2.3. Next-Generation Sequencing
2.4. Genome Annotation and Phylogenetic Analysis
3. Results
3.1. Characteristics of Patients with COVID-19 from the Southern Province of Zambia
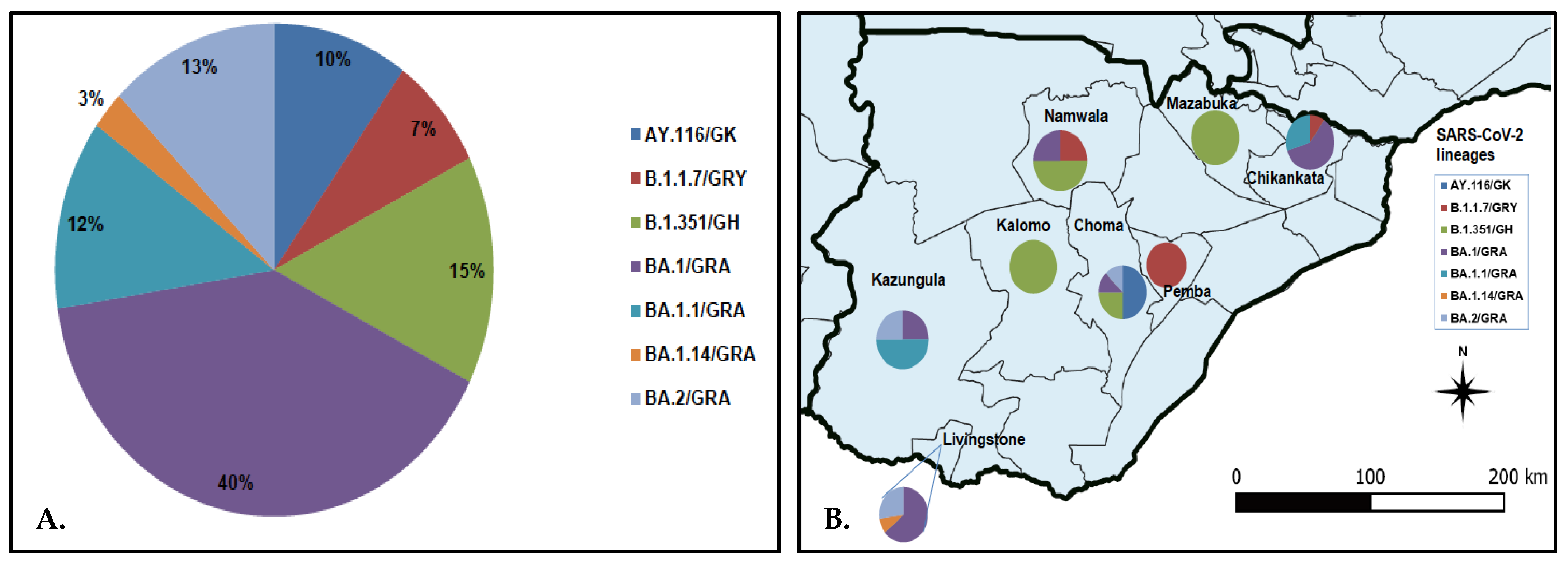
3.2. SARS-CoV-2 Lineage Assignment and Distribution in Southern Province
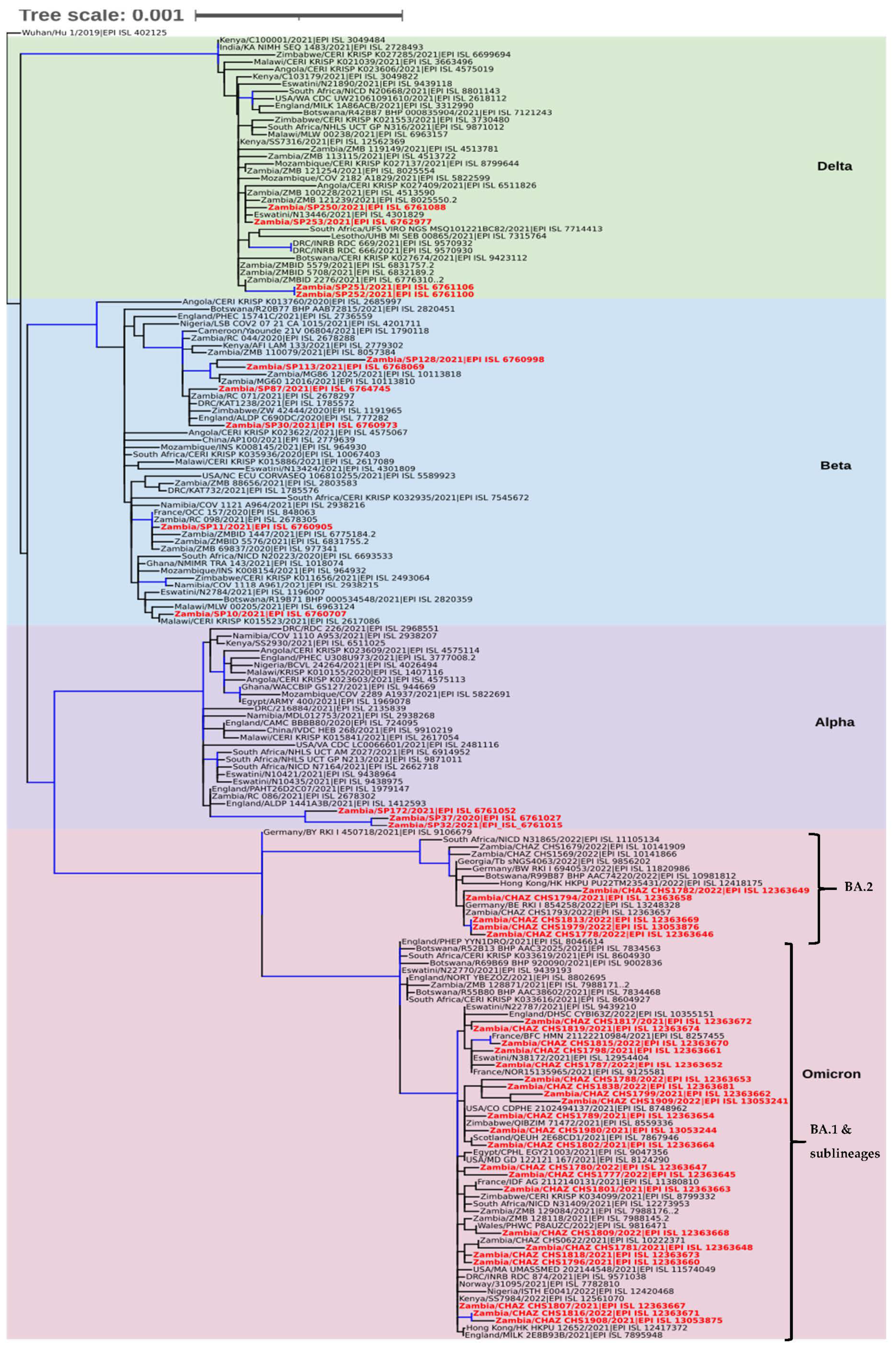
3.3. Phylogenetic Analysis
3.4. Molecular Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO Coronavirus (COVID-19) Dashboard. 2022. Available online: https://covid19.who.int/ (accessed on 7 July 2022).
- Uyoga, S.; Adetifa, I.M.O.; Karanja, H.K.; Nyagwange, J.; Tuju, J.; Wanjiku, P.; Aman, R.; Mwangangi, M.; Amoth, P.; Kasera, K.; et al. Seroprevalence of anti-SARS-CoV-2 IgG antibodies in Kenyan blood donors. Science 2021, 371, 79–82. [Google Scholar] [CrossRef]
- Mwananyanda, L.; Gill, C.J.; MacLeod, W.; Kwenda, G.; Pieciak, R.; Mupila, Z.; Lapidot, R.; Mupeta, F.; Forman, L.; Ziko, L.; et al. COVID-19 deaths in Africa: Prospective systematic postmortem surveillance study. BMJ 2021, 372, n334. [Google Scholar] [CrossRef]
- Chisale, M.R.O.; Ramazanu, S.; Mwale, S.E.; Kumwenda, P.; Chipeta, M.; Kaminga, A.C.; Nkhata, O.; Nyambalo, B.; Chavura, E.; Mbakaya, B.C. Seroprevalence of anti-SARS-CoV-2 antibodies in Africa: A systematic review and meta-analysis. Rev. Med. Virol. 2022, 32, e2271. [Google Scholar] [CrossRef]
- Gelanew, T.; Seyoum, B.; Mulu, A.; Mihret, A.; Abebe, M.; Wassie, L.; Gelaw, B.; Sorsa, A.; Merid, Y.; Muchie, Y.; et al. High seroprevalence of anti-SARS-CoV-2 antibodies among Ethiopian healthcare workers. BMC Infect. Dis. 2022, 22, 261. [Google Scholar] [CrossRef]
- Lewis, H.C.; Ware, H.; Whelan, M.G.; Subissi, L.; Li, Z.; Ma, X.; Nardone, A.; Valenciano, M.; Cheng, B.; Noel, K.C.; et al. SARS-CoV-2 infection in Africa: A systematic review and meta-analysis of standardised seroprevalence studies, from January 2020 to December 2021. medRxiv 2022, 2022.02.14.22270934. [Google Scholar] [CrossRef]
- World Health Organisation. Over Two-Thirds of Africans Exposed to Virus Which Causes COVID-19: WHO Study. 2022. Available online: https://www.afro.who.int/news/over-two-thirds-africans-exposed-virus-which-causes-covid-19-who-study (accessed on 1 June 2022).
- Massinga Loembé, M.; Tshangela, A.; Salyer, S.J.; Varma, J.K.; Ouma, A.E.O.; Nkengasong, J.N. COVID-19 in Africa: The spread and response. Nat. Med. 2020, 26, 999–1003. [Google Scholar] [CrossRef]
- Oluniyi, P. First African SARS-CoV-2 Genome Sequence from Nigerian COVID-19 Case: Virological.org; 2020. Available online: https://virological.org/t/first-african-sars-cov-2-genome-sequence-from-nigerian-covid-19-case/421 (accessed on 31 May 2022).
- World Health Organisation. A Second COVID-19 Case is Confirmed in Africa. 2020. Available online: https://www.afro.who.int/news/second-covid-19-case-confirmed-africa (accessed on 31 May 2022).
- Elimian, K.O.; Ochu, C.L.; Ilori, E.; Oladejo, J.; Igumbor, E.; Steinhardt, L.; Wagai, J.; Arinze, C.; Ukponu, W.; Obiekea, C.; et al. Descriptive epidemiology of coronavirus disease 2019 in Nigeria, 27 February–6 June 2020. Epidemiol. Infect. 2020, 148, e208. [Google Scholar] [CrossRef]
- Dufailu, O.A.; Afriyie-Asante, A.; Gyan, B.; Kwabena, D.A.; Yeboah, H.; Ntiakoh, F.; Asare-Werehene, M. COVID-19 in Africa: An ovarian victory? J. Ovarian Res. 2021, 14, 70. [Google Scholar] [CrossRef]
- Wilkinson, E.; Giovanetti, M.; Tegally, H.; San, J.E.; Lessells, R.; Cuadros, D.; Martin, D.P.; Rasmussen, D.A.; Zekri, A.R.N.; Sangare, A.K.; et al. A year of genomic surveillance reveals how the SARS-CoV-2 pandemic unfolded in Africa. Science 2021, 374, 423–431. [Google Scholar] [CrossRef]
- Sanyaolu, A.; Okorie, C.; Marinkovic, A.; Haider, N.; Abbasi, A.F.; Jaferi, U.; Prakash, S.; Balendra, V. The emerging SARS-CoV-2 variants of concern. Ther. Adv. Infect. Dis. 2021, 8, 20499361211024372. [Google Scholar] [CrossRef]
- Dinnon, K.H.; Leist, S.R.; Schäfer, A.; Edwards, C.E.; Martinez, D.R.; Montgomery, S.A.; West, A.; Yount, B.L., Jr.; Hou, Y.J.; Adams, L.E.; et al. A mouse-adapted model of SARS-CoV-2 to test COVID-19 countermeasures. Nature 2020, 586, 560–566. [Google Scholar] [CrossRef]
- Aleem, A.; Akbar Samad, A.B.; Slenker, A.K. Emerging Variants of SARS-CoV-2 And Novel Therapeutics Against Coronavirus (COVID-19). In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Ramanathan, M.; Ferguson, I.D.; Miao, W.; Khavari, P.A. SARS-CoV-2 B.1.1.7 and B.1.351 spike variants bind human ACE2 with increased affinity. Lancet Infect. Dis. 2021, 21, 1070. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; et al. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell 2020, 182, 1295–1310. [Google Scholar] [CrossRef]
- Holland, L.A.; Kaelin, E.A.; Maqsood, R.; Estifanos, B.; Wu, L.I.; Varsani, A.; Halden, R.U.; Hogue, B.G.; Scotch, M.; Lim, E.S. An 81-Nucleotide Deletion in SARS-CoV-2 ORF7a Identified from Sentinel Surveillance in Arizona (January to March 2020). J. Virol. 2020, 94, e00711-20. [Google Scholar] [CrossRef]
- Addetia, A.; Xie, H.; Roychoudhury, P.; Shrestha, L.; Loprieno, M.; Huang, M.-L.; Jerome, K.R.; Greninger, A.L. Identification of multiple large deletions in ORF7a resulting in in-frame gene fusions in clinical SARS-CoV-2 isolates. J. Clin. Virol. 2020, 129, 104523. [Google Scholar] [CrossRef]
- Mazur-Panasiuk, N.; Rabalski, L.; Gromowski, T.; Nowicki, G.; Kowalski, M.; Wydmanski, W.; Szulc, P.; Kosinski, M.; Gackowska, K.; Drweska-Matelska, N.; et al. Expansion of a SARS-CoV-2 Delta variant with an 872 nt deletion encompassing ORF7a, ORF7b and ORF8, Poland, July to August 2021. Eurosurveillance 2021, 26, 2100902. [Google Scholar] [CrossRef]
- Bal, A.; Destras, G.; Gaymard, A.; Bouscambert-Duchamp, M.; Valette, M.; Escuret, V.; Frobert, E.; Billaud, G.; Trouillet-Assant, S.; Cheynet, V.; et al. Molecular characterization of SARS-CoV-2 in the first COVID-19 cluster in France reveals an amino acid deletion in nsp2 (Asp268del). Clin. Microbiol. Infect. 2020, 26, 960–962. [Google Scholar] [CrossRef]
- Young, B.E.; Fong, S.-W.; Chan, Y.-H.; Mak, T.-M.; Ang, L.W.; Anderson, D.E.; Lee, C.Y.-P.; Amrun, S.N.; Lee, B.; Goh, Y.S.; et al. Effects of a major deletion in the SARS-CoV-2 genome on the severity of infection and the inflammatory response: An observational cohort study. Lancet 2020, 396, 603–611. [Google Scholar] [CrossRef]
- Volz, E.; Mishra, S.; Chand, M.; Barrett, J.C.; Johnson, R.; Geidelberg, L.; Hinsley, W.R.; Laydon, D.J.; Dabrera, G.; O’Toole, Á.; et al. Assessing transmissibility of SARS-CoV-2 lineage B.1.1.7 in England. Nature 2021, 593, 266–269. [Google Scholar] [CrossRef]
- Karadag, E. Increase in COVID-19 cases and case-fatality and case-recovery rates in Europe: A cross-temporal meta-analysis. J. Med. Virol. 2020, 92, 1511–1517. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef]
- Hirabara, S.M.; Serdan, T.D.A.; Gorjao, R.; Masi, L.N.; Pithon-Curi, T.C.; Covas, D.T.; Curi, R.; Durigon, E.L. SARS-CoV-2 Variants: Differences and potential of immune evasion. Front. Cell. Infect. Microbiol. 2022, 11, 781429. [Google Scholar] [CrossRef]
- Singanayagam, A.; Hakki, S.; Dunning, J.; Madon, K.J.; Crone, M.A.; Koycheva, A.; Derqui-Fernandez, N.; Barnett, J.L.; Whitfield, M.G.; Varro, R.; et al. Community transmission and viral load kinetics of the SARS-CoV-2 delta (B.1.617.2) variant in vaccinated and unvaccinated individuals in the UK: A prospective, longitudinal, cohort study. Lancet Infect. Dis. 2022, 22, 183–195. [Google Scholar] [CrossRef]
- Telenti, A.; Hodcroft, E.B.; Robertson, D.L. The evolution and biology of SARS-CoV-2 variants. Cold Spring Harb. Perspect. Med. 2022, 12, a041390. [Google Scholar] [CrossRef]
- Chipimo, P.J.; Barradas, D.T.; Kayeyi, N.; Zulu, P.M.; Muzala, K.; Mazaba, M.L.; Hamoonga, R.; Musonda, K.; Monze, M.; Kapata, N.; et al. First 100 persons with COVID-19-Zambia, March 18–April 28, 2020. MMWR. Morb. Mortal. Wkly. Rep. 2020, 69, 1547–1548. [Google Scholar] [CrossRef]
- Zambia National Public Health Institute. Zambia COVID-19 Dashboard. 2022. Available online: https://www.arcgis.com/apps/dashboards/3b3a01c1d8444932ba075fb44b119b63 (accessed on 21 May 2022).
- Worldometer. Coronavirus. Available online: https://www.worldometers.info/coronavirus/country/zambia/ (accessed on 13 June 2022).
- Pandemic Coronavirus Causing COVID-19. 2020. Available online: https://www.epicov.org/epi3/frontend#1739f3 (accessed on 21 May 2022).
- Simulundu, E.; Mupeta, F.; Chanda-Kapata, P.; Saasa, N.; Changula, K.; Muleya, W.; Chitanga, S.; Mwanza, M.; Simusika, P.; Chambaro, H.; et al. First COVID-19 case in Zambia-Comparative phylogenomic analyses of SARS-CoV-2 detected in African countries. Int. J. Infect. Dis. 2021, 102, 455–459. [Google Scholar] [CrossRef]
- Mwenda, M.; Saasa, N.; Sinyange, N.; Busby, G.; Chipimo, P.J.; Hendry, J.; Kapona, O.; Yingst, S.; Hines, J.Z.; Minchella, P.; et al. Detection of B.1.351 SARS-CoV-2 Variant Strain-Zambia, December 2020. MMWR. Morb. Mortal. Wkly. Rep. 2021, 70, 280–282. [Google Scholar] [CrossRef]
- Lu, X.; Wang, L.; Sakthivel, S.K.; Whitaker, B.; Murray, J.; Kamili, S.; Lynch, B.; Malapati, L.; Burke, S.A.; Harcourt, J.; et al. US CDC real-time reverse transcription PCR panel for detection of severe acute respiratory syndrome coronavirus 2. Emerg. Infect. Dis. 2020, 26, 1654. [Google Scholar] [CrossRef]
- Quick, J. nCoV-2019 Sequencing Protocol v3 (LoCost). Protocols.io. 2020. Available online: https://protocols.io/view/ncov-2019-sequencing-protocol-v3-locost-bh42j8ye (accessed on 5 April 2021).
- Tyson, J.R.; James, P.; Stoddart, D.; Sparks, N.; Wickenhagen, A.; Hall, G.; Choi, J.H.; Lapointe, H.; Kamelian, K.; Smith, A.D.; et al. Improvements to the ARTIC multiplex PCR method for SARS-CoV-2 genome sequencing using nanopore. bioRxiv 2020, 2020.09.04.283077. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Briefings Bioinform. 2017, 20, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Lethiec, F.; Duroux, P.; Gascuel, O. PHYML Online—a web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Res. 2005, 33, W557–W559. [Google Scholar] [CrossRef] [PubMed]
- Lefort, V.; Longueville, J.-E.; Gascuel, O. SMS: Smart model selection in PhyML. Mol. Biol. Evol. 2017, 34, 2422–2424. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Taboada, B.; Zárate, S.; Iša, P.; Boukadida, C.; Vazquez-Perez, J.A.; Muñoz-Medina, J.E.; Ramírez-González, J.E.; Comas-García, A.; Grajales-Muñiz, C.; Rincón-Rubio, A.; et al. Genetic Analysis of SARS-CoV-2 Variants in Mexico during the First Year of the COVID-19 Pandemic. Viruses 2021, 13, 2161. [Google Scholar] [CrossRef]
- Jin, J.M.; Bai, P.; He, W.; Wu, F.; Liu, X.F.; Han, D.M.; Liu, S.; Yang, J.K. Gender differences in patients with COVID-19: Focus on severity and mortality. Front. Public Health 2020, 8, 152. [Google Scholar] [CrossRef]
- Yadav, P.D.; Nyayanit, D.A.; Majumdar, T.; Patil, S.; Kaur, H.; Gupta, N.; Shete, A.M.; Pandit, P.; Kumar, A.; Aggarwal, N.; et al. An epidemiological analysis of SARS-CoV-2 genomic sequences from different regions of India. Viruses 2021, 13, 925. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Lessells, R.J.; Giandhari, J.; Pillay, S.; Msomi, N.; Mlisana, K.; Bhiman, J.N.; von Gottberg, A.; Walaza, S.; et al. Sixteen novel lineages of SARS-CoV-2 in South Africa. Nat. Med. 2021, 27, 440–446. [Google Scholar] [CrossRef]
- Viana, R.; Moyo, S.; Amoako, D.G.; Tegally, H.; Scheepers, C.; Althaus, C.L.; Anyaneji, U.J.; Bester, P.A.; Boni, M.F.; Chand, M.; et al. Rapid epidemic expansion of the SARS-CoV-2 Omicron variant in southern Africa. Nature 2022, 603, 679–686. [Google Scholar] [CrossRef]
- Salyer, S.J.; Maeda, J.; Sembuche, S.; Kebede, Y.; Tshangela, A.; Moussif, M.; Ihekweazu, C.; Mayet, N.; Abate, E.; Ouma, A.O.; et al. The first and second waves of the COVID-19 pandemic in Africa: A cross-sectional study. Lancet 2021, 397, 1265–1275. [Google Scholar] [CrossRef]
- Plante, J.A.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R.; et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature 2021, 592, 116–121. [Google Scholar] [CrossRef]
- Khan, K.; Karim, F.; Ganga, Y.; Bernstein, M.; Jule, Z.; Reedoy, K.; Cele, S.; Lustig, G.; Amoako, D.; Wolter, N.; et al. Omicron BA.4/BA.5 escape neutralizing immunity elicited by BA.1 infection. Nat. Commun. 2022, 13, 4686. [Google Scholar] [CrossRef]
- Tegally, H.; Moir, M.; Everatt, J.; Giovanetti, M.; Scheepers, C.; Wilkinson, E.; Subramoney, K.; Moyo, S.; Amoako, D.G.; Althaus, C.L.; et al. Continued emergence and evolution of omicron in South Africa: New BA.4 and BA.5 lineages. medRxiv 2022, 2022.05.01.22274406. [Google Scholar] [CrossRef]
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020, 17, 613–620. [Google Scholar] [CrossRef]
- Du, L.; He, Y.; Zhou, Y.; Liu, S.; Zheng, B.-J.; Jiang, S. The spike protein of SARS-CoV—a target for vaccine and therapeutic development. Nat. Rev. Microbiol. 2009, 7, 226–236. [Google Scholar] [CrossRef]
- Lu, L.; Liu, Q.; Zhu, Y.; Chan, K.-H.; Qin, L.; Li, Y.; Wang, Q.; Chan, J.F.-W.; Du, L.; Yu, F.; et al. Structure-based discovery of Middle East respiratory syndrome coronavirus fusion inhibitor. Nat. Commun. 2014, 5, 3067. [Google Scholar] [CrossRef]
- Du, L.; Tai, W.; Yang, Y.; Zhao, G.; Zhu, Q.; Sun, S.; Liu, C.; Tao, X.; Tseng, X.T.C.-T.K.; Perlman, S.; et al. Introduction of neutralizing immunogenicity index to the rational design of MERS coronavirus subunit vaccines. Nat. Commun. 2016, 7, 13473. [Google Scholar] [CrossRef]
- Duan, L.; Zheng, Q.; Zhang, H.; Niu, Y.; Lou, Y.; Wang, H. The SARS-CoV-2 spike glycoprotein biosynthesis, structure, function, and antigenicity: Implications for the design of spike-based vaccine immunogens. Front. Immunol. 2020, 11, 576622. [Google Scholar] [CrossRef]
- Sahin, E.; Bozdayi, G.; Yigit, S.; Muftah, H.; Dizbay, M.; Tunccan, O.G.; Fidan, I.; Caglar, K. Genomic characterization of SARS-CoV-2 isolates from patients in Turkey reveals the presence of novel mutations in spike and nsp12 proteins. J. Med. Virol. 2021, 93, 6016–6026. [Google Scholar] [CrossRef]
- Al-Mahruqi, S.; Al-Wahaibi, A.; Khan, A.L.; Al-Jardani, A.; Asaf, S.; Alkindi, H.; Al-Kharusi, S.; Al-Rawahi, A.N.; Al-Salmani, M.; Al-Shukri, I.; et al. Molecular epidemiology of COVID-19 in Oman: A molecular and surveillance study for the early transmission of COVID-19 in the country. Int. J. Infect. Dis. 2021, 104, 139–149. [Google Scholar] [CrossRef]
- Zekri, A.-R.N.; Amer, K.E.; Hafez, M.M.; Hassan, Z.K.; Ahmed, O.S.; Soliman, H.K.; Bahnasy, A.A.; Hamid, W.A.; Gad, A.; Ali, M.; et al. Genomic characterization of SARS-CoV-2 in Egypt. J. Adv. Res. 2021, 30, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Agoti, C.N.; Githinji, G.; Mohammed, K.S.; Lambisia, A.W.; de Laurent, Z.R.; Mburu, M.W.; Ong’Era, E.M.; Morobe, J.M.; Otieno, E.; Azali, H.A.; et al. Detection of SARS-CoV-2 variant 501Y.V2 in comoros islands in January 2021. Wellcome Open Res. 2021, 6, 192. [Google Scholar] [CrossRef]
- Obeid, D.A.; Alsanea, M.S.; Alnemari, R.T.; Al-Qahtani, A.A.; Althawadi, S.I.; Mutabagani, M.S.; Almaghrabi, R.S.; Alhadheq, F.M.; Alahideb, B.M.; Alhamlan, F.S. SARS-CoV-2 genetic diversity and variants of concern in Saudi Arabia. J. Infect. Dev. Ctries. 2021, 15, 1782–1791. [Google Scholar] [CrossRef]
- Flores-Alanis, A.; Cruz-Rangel, A.; Rodríguez-Gómez, F.; González, J.; Torres-Guerrero, C.A.; Delgado, G.; Cravioto, A.; Morales-Espinosa, R. Molecular epidemiology surveillance of SARS-CoV-2: Mutations and genetic diversity one year after emerging. Pathogens 2021, 10, 184. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Kader, S.B.; Rizvi, S.M.S. Molecular characterization of SARS-CoV-2 from Bangladesh: Implications in genetic diversity, possible origin of the virus, and functional significance of the mutations. Heliyon 2021, 7, e07866. [Google Scholar] [CrossRef] [PubMed]
- Eskier, D.; Karakülah, G.; Suner, A.; Oktay, Y. RdRp mutations are associated with SARS-CoV-2 genome evolution. PeerJ 2020, 8, e9587. [Google Scholar] [CrossRef]
- Haddad, D.; John, S.E.; Mohammad, A.; Hammad, M.M.; Hebbar, P.; Channanath, A.; Nizam, R.; Al-Qabandi, S.; Al Madhoun, A.; Alshukry, A.; et al. SARS-CoV-2: Possible recombination and emergence of potentially more virulent strains. PLoS ONE 2021, 16, e0251368. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G increases infectivity of the COVID-19 virus. Cell 2020, 182, 812–827. [Google Scholar] [CrossRef]
- Gómez, C.E.; Perdiguero, B.; Esteban, M. Emerging SARS-CoV-2 Variants and Impact in Global Vaccination Programs against SARS-CoV-2/COVID-19. Vaccines 2021, 9, 243. [Google Scholar] [CrossRef]
- Kang, N.; Prevention, N.G.C.F.D.C.A.; Liu, Y.; Bi, F.; Wang, J.; Chen, H.; Liang, Z.; Chen, M.; Wei, G.; Li, A.; et al. Detection of variants of B.1.617 lineage from five returning Chinese nationals at a Guangxi-vietnam border port—Guangxi Zhuang Autonomous Region, China. China CDC Wkly. 2021, 30, 653–655. [Google Scholar] [CrossRef]
- Malune, P.; Piras, G.; Monne, M.; Fiamma, M.; Asproni, R.; Fancello, T.; Manai, A.; Carta, F.; Fancello, P.; Rosu, V.; et al. Molecular characterization of severe acute respiratory syndrome coronavirus 2 isolates from central inner sardinia. Front. Microbiol. 2022, 12, 827799. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.K.; Kumar, S.; Ansari, S.; Paweska, J.T.; Maurya, V.K.; Tripathi, A.K.; Abdel-Moneim, A.S. Characterization of the novel SARS-CoV-2 Omicron (B.1.1.529) variant of concern and its global perspective. J. Med. Virol. 2022, 94, 1738–1744. [Google Scholar] [CrossRef] [PubMed]
- Geddes, L. From Alpha to Omicron: Everything You Need to Know about Coronavirus Variants of Concern: Gavi the Vaccine Alliance. 2021. Available online: https://www.gavi.org/vaccineswork/alpha-omicron-%20everything-you-need-know-about-coronavirus-variants-concern (accessed on 24 May 2022).

{kind=link}
{kind=link}
{kind=link}
| Parameters | Sample Distribution n (%), Overall, n = 198 |
|---|---|
| Age Group | |
| 0–14 Years | 7 (3.5) |
| 15–50 Years | 171 (86.4) |
| >50 Years | 15 (7.6) |
| Unknown | 5 (2.5) |
| Gender | |
| Female | 104 (52.5) |
| Male | 93 (47.0) |
| Unknown | 1 (0.5) |
| Genome Segment | Missense Mutation | Synonymous Mutation | Deletion | Insertion | Others | Total Mutation |
|---|---|---|---|---|---|---|
| Coding Region | ||||||
| ORF1ab | 74 | 48 | 9 | 3 | 0 | 134 |
| Spike | 65 | 3 | 10 | 4 | 0 | 82 |
| ORF3a | 5 | 4 | 0 | 0 | 0 | 9 |
| Envelope | 5 | 0 | 0 | 0 | 0 | 5 |
| Membrane | 5 | 2 | 0 | 0 | 0 | 7 |
| ORF6 | 2 | 2 | 0 | 0 | 0 | 4 |
| ORF7a | 2 | 0 | 0 | 0 | 0 | 2 |
| ORF7b | 4 | 1 | 2 | 2 | 0 | 9 |
| ORF8 | 4 | 3 | 1 | 0 | 1 1 | 9 |
| Nucleocapsid | 16 | 3 | 1 | 0 | 0 | 20 |
| Non-coding Region 2 | ||||||
| 5′UTR | 0 | 0 | 0 | 0 | 4 | 4 |
| 3′UTR | 0 | 0 | 0 | 0 | 7 | 7 |
| Total | 182 | 66 | 23 | 9 | 12 | 292 |
| SARS-CoV-2 Variants | Spike Mutations 1 |
|---|---|
| Wuhan-Hu-1 (wild-type) | - |
| Alpha (B.1.1.7) | ∆H69, ∆Y145, N501Y, A570D, D614G, P681H, T716I, T874I, S982A, D1118H |
| Beta (B.1.351) | L18F, D80A, D215G, ∆L242, T307P, N317F, S325P, I326K, V327A, K417N, E484K, N501Y, D614G, A701V, A1087S |
| Delta (AY.116) | T19R, T95I, G142D, ∆E156, L452R, T478K, D614G, P681R, D950N |
| Omicron (BA.1, BA.1.1, BA.1.14, BA.2) | T19I, ∆L24, ∆A67, ∆A67, ∆I68, T95I, ∆G142, G142D, V193L, Y200C, insI210, ∆N211, N211K, L212C, V213G, insS214, insV213, insR214, insV213, insR214, R214R, A243S, L244S, G339D, R346K, Y369Y, S371L, S371F, S373P, S375F, T376A, D405N, R408S, K417N, N440K, G446S, T470A, S477N, T478K, E484A, Q493R, G496S, Q498R, N501Y, Y505H, T547K, D614G, H655Y, N679K, P681H, N764K, D796Y, N856K, Q954H, N969K, L981F, V1104L, D1127G, D1146D, V1264L |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Katowa, B.; Kalonda, A.; Mubemba, B.; Matoba, J.; Shempela, D.M.; Sikalima, J.; Kabungo, B.; Changula, K.; Chitanga, S.; Kasonde, M.; et al. Genomic Surveillance of SARS-CoV-2 in the Southern Province of Zambia: Detection and Characterization of Alpha, Beta, Delta, and Omicron Variants of Concern. Viruses 2022, 14, 1865. https://doi.org/10.3390/v14091865
Katowa B, Kalonda A, Mubemba B, Matoba J, Shempela DM, Sikalima J, Kabungo B, Changula K, Chitanga S, Kasonde M, et al. Genomic Surveillance of SARS-CoV-2 in the Southern Province of Zambia: Detection and Characterization of Alpha, Beta, Delta, and Omicron Variants of Concern. Viruses. 2022; 14(9):1865. https://doi.org/10.3390/v14091865
Chicago/Turabian StyleKatowa, Ben, Annie Kalonda, Benjamin Mubemba, Japhet Matoba, Doreen Mainza Shempela, Jay Sikalima, Boniface Kabungo, Katendi Changula, Simbarashe Chitanga, Mpanga Kasonde, and et al. 2022. "Genomic Surveillance of SARS-CoV-2 in the Southern Province of Zambia: Detection and Characterization of Alpha, Beta, Delta, and Omicron Variants of Concern" Viruses 14, no. 9: 1865. https://doi.org/10.3390/v14091865
APA StyleKatowa, B., Kalonda, A., Mubemba, B., Matoba, J., Shempela, D. M., Sikalima, J., Kabungo, B., Changula, K., Chitanga, S., Kasonde, M., Kapona, O., Kapata, N., Musonda, K., Monze, M., Tembo, J., Bates, M., Zumla, A., Sutcliffe, C. G., Kajihara, M., ... Simulundu, E. (2022). Genomic Surveillance of SARS-CoV-2 in the Southern Province of Zambia: Detection and Characterization of Alpha, Beta, Delta, and Omicron Variants of Concern. Viruses, 14(9), 1865. https://doi.org/10.3390/v14091865