
Molecular Epidemiology and Evolutionary Analysis of Avian Influenza A(H5) Viruses Circulating in Egypt, 2019–2021

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples Collection
2.2. Molecular Detection and Virus Isolation
2.3. Sequencing and Phylogenetic Analyses
2.4. Times of Most Recent Common Ancestor (tMRCAs)
2.5. Selection Pressure
3. Results
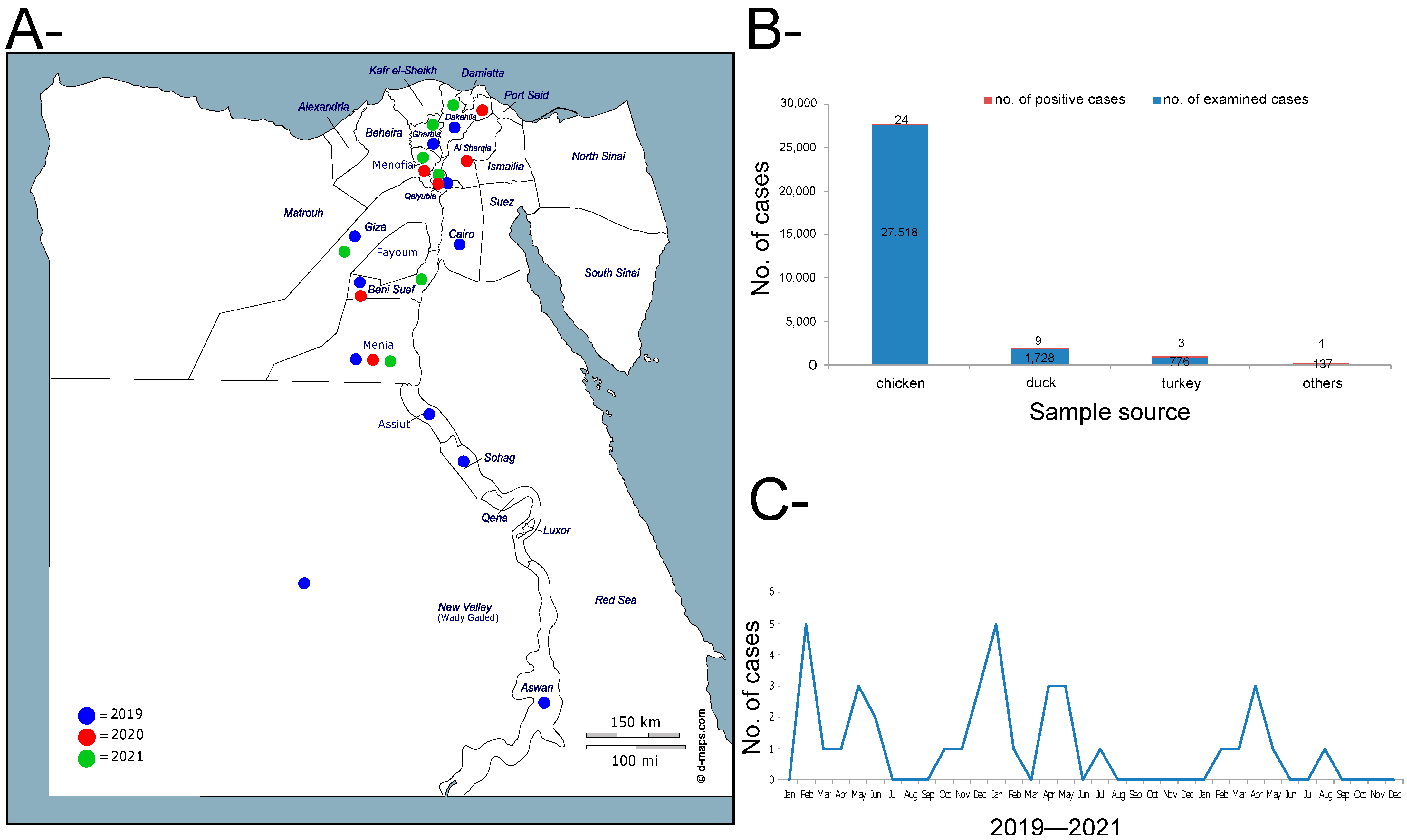
3.1. Active and Passive Surveillance
3.2. Genetic Diversity and Selection Pressure
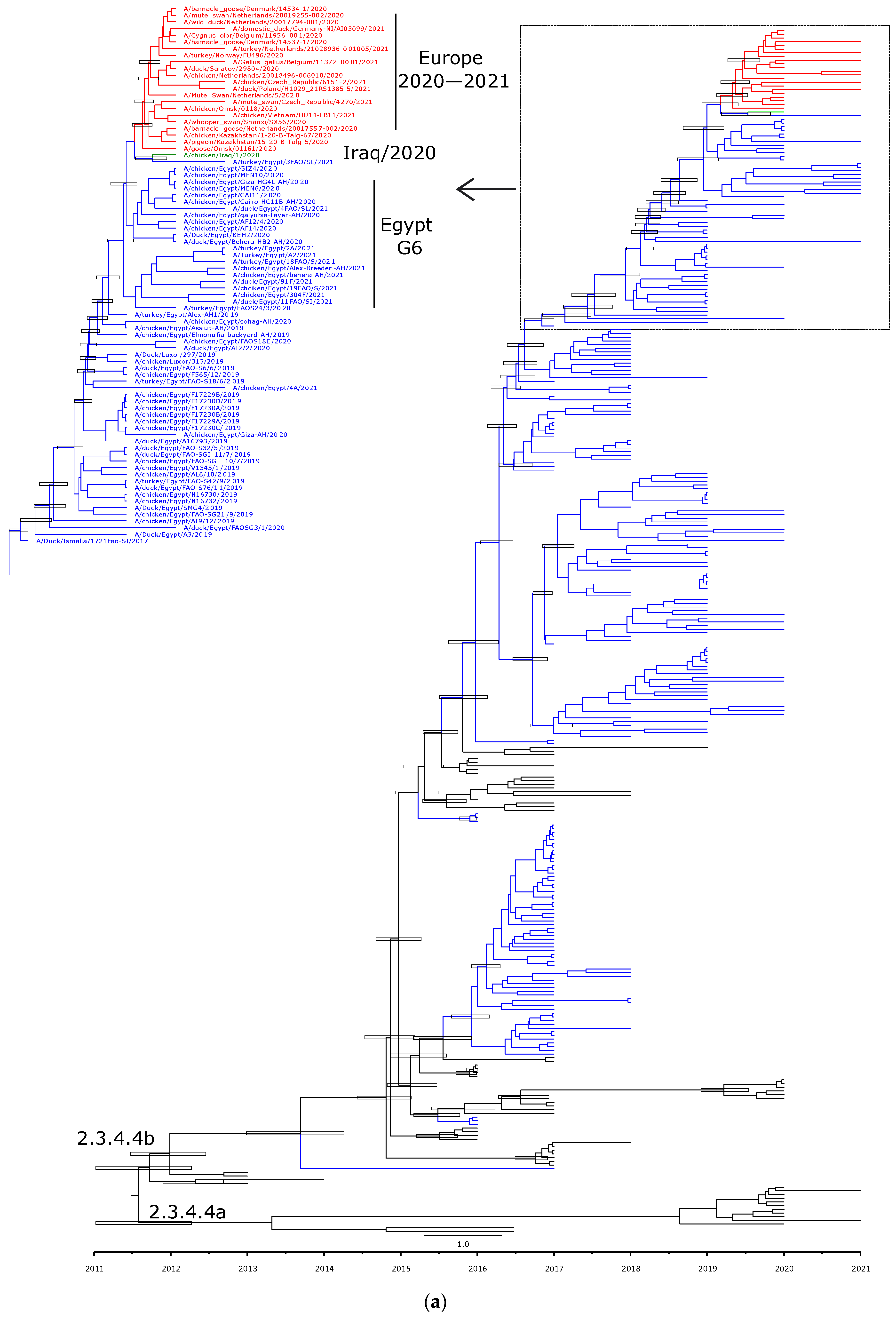
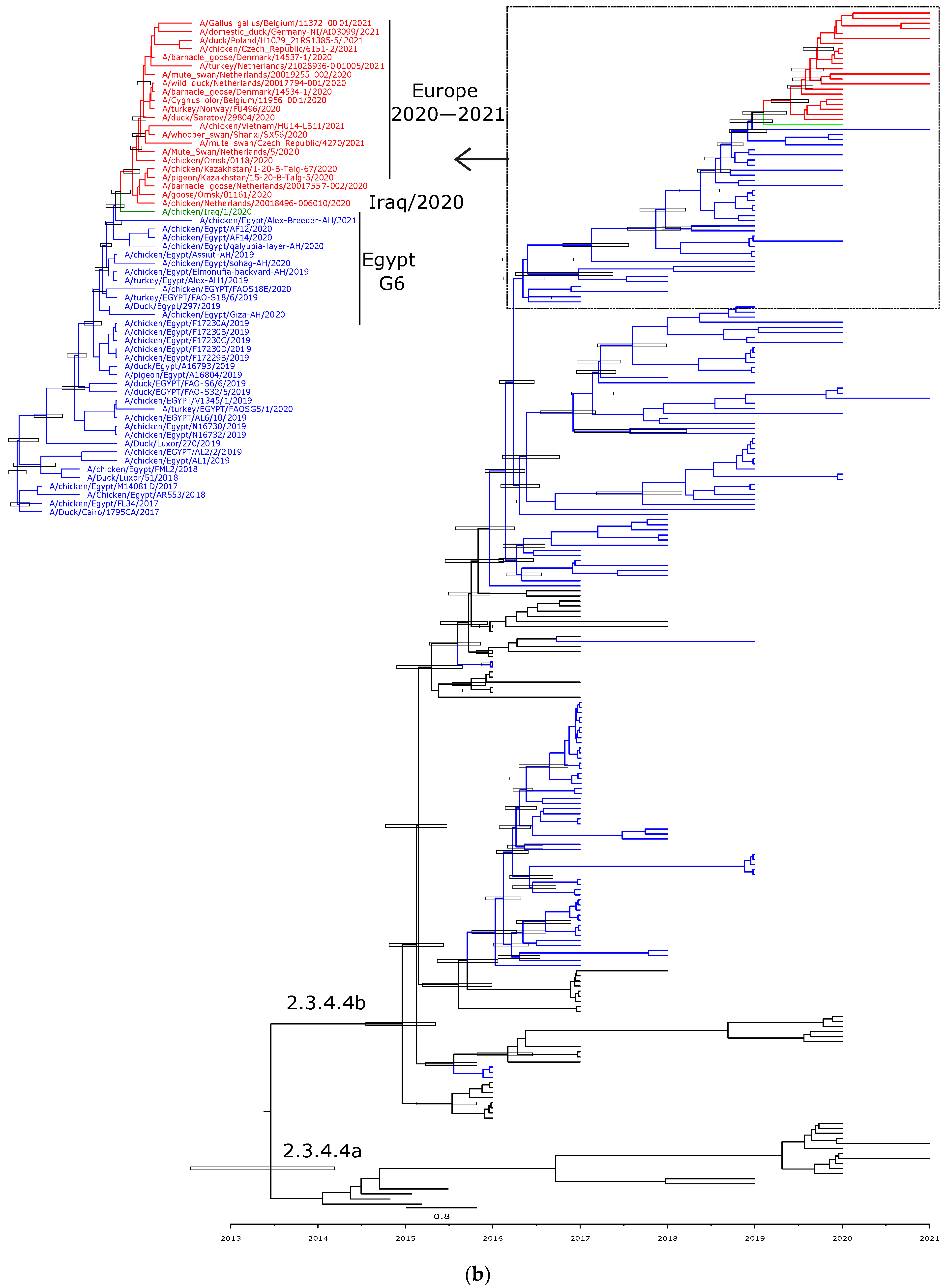
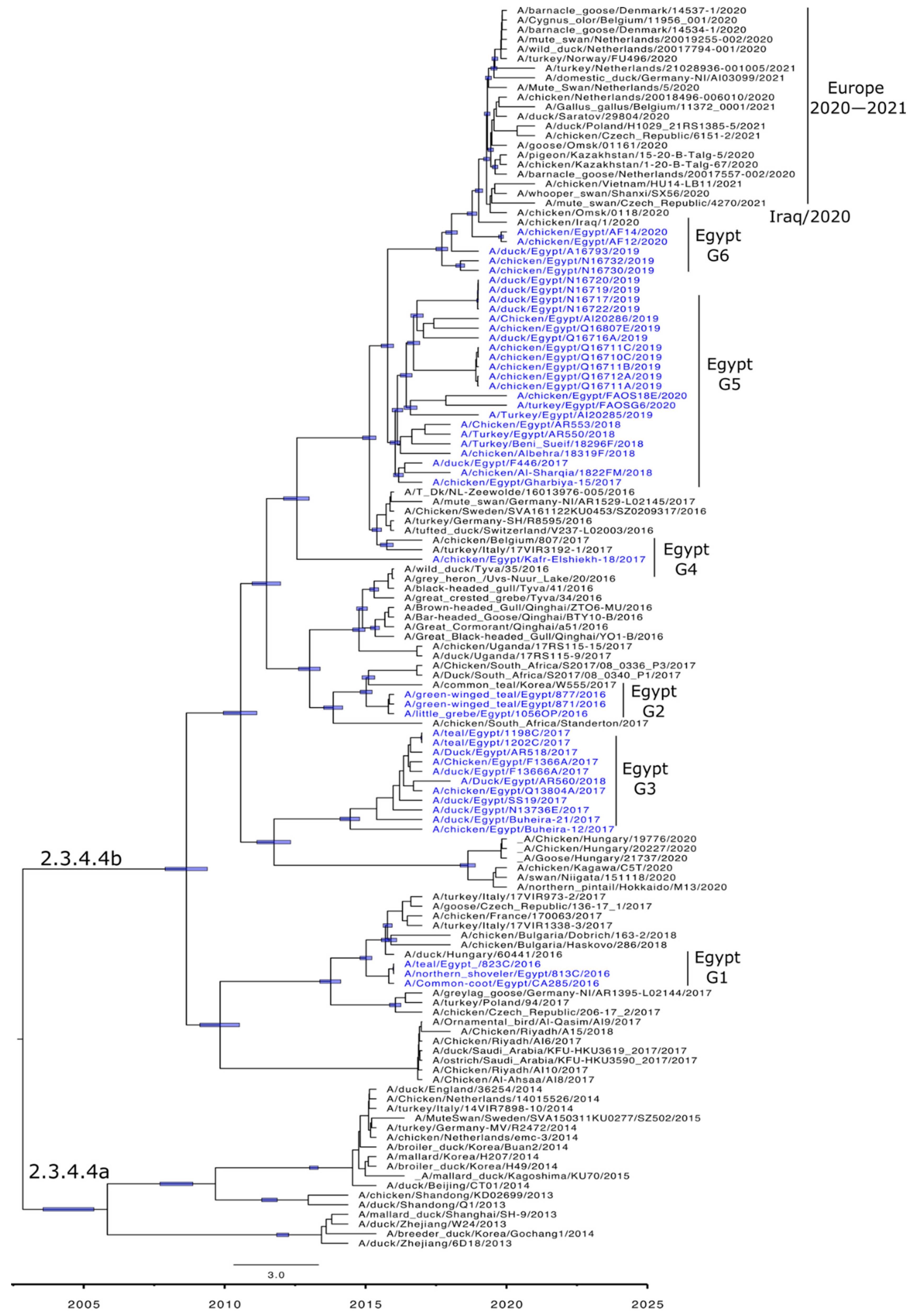
3.3. Phylogenetic Characterization and Molecular Dating
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nuñez, I.A.; Ross, T.M. A review of H5Nx avian influenza viruses. Ther. Adv. Vaccines Immunother. 2019, 7, 2515135518821625. [Google Scholar] [CrossRef]
- Pohlmann, A.; King, J.; Fusaro, A.; Zecchin, B.; Banyard, A.C.; Brown, I.H.; Byrne, A.M.P.; Beerens, N.; Liang, Y.; Heutink, R.; et al. Has Epizootic Become Enzootic? Evidence for a Fundamental Change in the Infection Dynamics of Highly Pathogenic Avian Influenza in Europe, 2021. mBio 2022. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Gu, M.; Zhong, L.; Duan, Z.; Zhang, Y.; Zhu, Y.; Zhao, G.; Zhao, M.; Chen, Z.; Hu, S.; et al. Characterization of three H5N5 and one H5N8 highly pathogenic avian influenza viruses in China. Vet. Microbiol. 2013, 163, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-J.; Kang, H.-M.; Lee, E.-K.; Song, B.-M.; Jeong, J.; Kwon, Y.-K.; Kim, H.-R.; Lee, K.-J.; Hong, M.-S.; Jang, I.; et al. Novel Reassortant Influenza A(H5N8) Viruses, South Korea, 2014. Emerg. Infect. Dis. 2014, 20, 1087–1089. [Google Scholar] [CrossRef] [PubMed]
- Bevins, S.N.; Dusek, R.J.; White, C.L.; Gidlewski, T.; Bodenstein, B.; Mansfield, K.G.; DeBruyn, P.; Kraege, D.; Rowan, E.; Gillin, C.; et al. Widespread detection of highly pathogenic H5 influenza viruses in wild birds from the Pacific Flyway of the United States. Sci. Rep. 2016, 6, 28980. [Google Scholar] [CrossRef]
- Globig, A.; Staubach, C.; Sauter-Louis, C.; Dietze, K.; Homeier-Bachmann, T.; Probst, C.; Gethmann, J.; Depner, K.R.; Grund, C.; Harder, T.C.; et al. Highly Pathogenic Avian Influenza H5N8 Clade 2.3.4.4b in Germany in 2016/2017. Front. Vet. Sci. 2017, 4, 240. [Google Scholar] [CrossRef] [PubMed]
- Lewis, N.S.; Banyard, A.C.; Whittard, E.; Karibayev, T.; Al Kafagi, T.; Chvala, I.; Byrne, A.; Meruyert Akberovna, S.; King, J.; Harder, T.; et al. Emergence and spread of novel H5N8, H5N5 and H5N1 clade 2.3.4.4 highly pathogenic avian influenza in 2020. Emerg. Microbes Infect. 2021, 10, 148–151. [Google Scholar] [CrossRef] [PubMed]
- Lycett, S.J.; Pohlmann, A.; Staubach, C.; Caliendo, V.; Woolhouse, M.; Beer, M.; Kuiken, T. Genesis and spread of multiple reassortants during the 2016/2017 H5 avian influenza epidemic in Eurasia. Proc. Natl. Acad. Sci. USA 2020, 117, 20814–20825. [Google Scholar] [CrossRef] [PubMed]
- Napp, S.; Majó, N.; Sánchez-Gónzalez, R.; Vergara-Alert, J. Emergence and spread of highly pathogenic avian influenza A(H5N8) in Europe in 2016-2017. Transbound. Emerg. Dis. 2018, 65, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Fusaro, A.; Zecchin, B.; Vrancken, B.; Abolnik, C.; Ademun, R.; Alassane, A.; Arafa, A.; Awuni, J.A.; Couacy-Hymann, E.; Coulibaly, M.B.; et al. Disentangling the role of Africa in the global spread of H5 highly pathogenic avian influenza. Nat. Commun. 2019, 10, 5310. [Google Scholar] [CrossRef] [PubMed]
- WHO. Human Infection with Avian Influenza A (H5N8)—The Russian Federation; World Health Organisation: Geneva, Switzerland, 2021; Available online: https://www.who.int/csr/don/26-feb-2021-influenza-a-russian-federation/en/ (accessed on 2 March 2021).
- Selim, A.A.; Erfan, A.M.; Hagag, N.; Zanaty, A.; Samir, A.H.; Samy, M.; Abdelhalim, A.; Arafa, A.A.; Soliman, M.A.; Shaheen, M.; et al. Highly Pathogenic Avian Influenza Virus (H5N8) Clade 2.3.4.4 Infection in Migratory Birds, Egypt. Emerg. Infect. Dis. 2017, 23, 1048–1051. [Google Scholar] [CrossRef] [PubMed]
- Yehia, N.; Naguib, M.M.; Li, R.; Hagag, N.; El-Husseiny, M.; Mosaad, Z.; Nour, A.; Rabea, N.; Hasan, W.M.; Hassan, M.K.; et al. Multiple introductions of reassorted highly pathogenic avian influenza viruses (H5N8) clade 2.3.4.4b causing outbreaks in wild birds and poultry in Egypt. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2018, 58, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Hassan, K.E.; Saad, N.; Abozeid, H.H.; Shany, S.; El-Kady, M.F.; Arafa, A.; El-Sawah, A.A.A.; Pfaff, F.; Hafez, H.M.; Beer, M.; et al. Genotyping and reassortment analysis of highly pathogenic avian influenza viruses H5N8 and H5N2 from Egypt reveals successive annual replacement of genotypes. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2020, 84, 104375. [Google Scholar] [CrossRef]
- Yehia, N.; Hassan, W.M.M.; Sedeek, A.; Elhusseiny, M.H. Genetic variability of avian influenza virus subtype H5N8 in Egypt in 2017 and 2018. Arch. Virol. 2020, 165, 1357–1366. [Google Scholar] [CrossRef] [PubMed]
- Salaheldin, A.H.; El-Hamid, H.S.; Elbestawy, A.R.; Veits, J.; Hafez, H.M.; Mettenleiter, T.C.; Abdelwhab, E.M. Multiple Introductions of Influenza A(H5N8) Virus into Poultry, Egypt, 2017. Emerg. Infect. Dis. 2018, 24, 943–946. [Google Scholar] [CrossRef] [PubMed]
- Moatasim, Y.; Kandeil, A.; Aboulhoda, B.E.; El-Shesheny, R.; Alkhazindar, M.; AbdElSalam, E.T.; Kutkat, O.; Kamel, M.N.; El Taweel, A.N.; Mostafa, A.; et al. Comparative Virological and Pathogenic Characteristics of Avian Influenza H5N8 Viruses Detected in Wild Birds and Domestic Poultry in Egypt during the Winter of 2016/2017. Viruses 2019, 11, 990. [Google Scholar] [CrossRef] [PubMed]
- Hagag, N.M.; Erfan, A.M.; El-Husseiny, M.; Shalaby, A.G.; Saif, M.A.; Tawakol, M.M.; Nour, A.A.; Selim, A.A.; Arafa, A.S.; Hassan, M.K.; et al. Isolation of a Novel Reassortant Highly Pathogenic Avian Influenza (H5N2) Virus in Egypt. Viruses 2019, 11, 565. [Google Scholar] [CrossRef]
- Hassan, K.E.; King, J.; El-Kady, M.; Afifi, M.; Abozeid, H.H.; Pohlmann, A.; Beer, M.; Harder, T. Novel Reassortant Highly Pathogenic Avian Influenza A(H5N2) Virus in Broiler Chickens, Egypt. Emerg. Infect. Dis. 2020, 26, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Beerens, N.; Heutink, R.; Harders, F.; Roose, M.; Pritz-Verschuren, S.B.E.; Germeraad, E.A.; Engelsma, M. Incursion of Novel Highly Pathogenic Avian Influenza A(H5N8) Virus, the Netherlands, October 2020. Emerg. Infect. Dis. 2021, 27, 1750–1753. [Google Scholar] [CrossRef] [PubMed]
- Amer, F.; Li, R.; Rabie, N.; El-Husseiny, M.H.; Yehia, N.; Hagag, N.M.; Samy, M.; Selim, A.; Hassan, M.K.; Hassan, W.M.M.; et al. Temporal Dynamics of Influenza A(H5N1) Subtype before and after the Emergence of H5N8. Viruses 2021, 13, 1565. [Google Scholar] [CrossRef]
- Kandeil, A.; Hicks, J.T.; Young, S.G.; El Taweel, A.N.; Kayed, A.S.; Moatasim, Y.; Kutkat, O.; Bagato, O.; McKenzie, P.P.; Cai, Z.; et al. Active surveillance and genetic evolution of avian influenza viruses in Egypt, 2016-2018. Emerg. Microbes Infect. 2019, 8, 1370–1382. [Google Scholar] [CrossRef] [PubMed]
- Hassan, K.E.; El-Kady, M.F.; El-Sawah, A.A.A.; Luttermann, C.; Parvin, R.; Shany, S.; Beer, M.; Harder, T. Respiratory disease due to mixed viral infections in poultry flocks in Egypt between 2017 and 2018: Upsurge of highly pathogenic avian influenza virus subtype H5N8 since 2018. Transbound. Emerg. Dis. 2019, 68, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Spackman, E.; Senne, D.A.; Bulaga, L.L.; Myers, T.J.; Perdue, M.L.; Garber, L.P.; Lohman, K.; Daum, L.T.; Suarez, D.L. Development of real-time RT-PCR for the detection of avian influenza virus. Avian Dis. 2003, 47, 1079–1082. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, B.; Hoffmann, D.; Henritzi, D.; Beer, M.; Harder, T.C. Riems influenza a typing array (RITA): An RT-qPCR-based low density array for subtyping avian and mammalian influenza a viruses. Sci. Rep. 2016, 6, 27211. [Google Scholar] [CrossRef]
- Singapore Tourism Board. Chapter 2.3.4. Avian Influenza. Available online: http://www.oie.int/fileadmin/Home/eng/Health_standards/tahm/2.03.04_AI.pdf (accessed on 22 November 2014).
- Hoper, D.; Hoffmann, B.; Beer, M. Simple, sensitive, and swift sequencing of complete H5N1 avian influenza virus genomes. J. Clin. Microbiol. 2009, 47, 674–679. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V.; Kosakovsky Pond, S.L. Datamonkey 2.0: A Modern Web Application for Characterizing Selective and Other Evolutionary Processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar] [CrossRef]
- Cai, Z.; Ducatez, M.F.; Yang, J.; Zhang, T.; Long, L.P.; Boon, A.C.; Webby, R.J.; Wan, X.F. Identifying antigenicity-associated sites in highly pathogenic H5N1 influenza virus hemagglutinin by using sparse learning. J. Mol. Biol. 2012, 422, 145–155. [Google Scholar] [CrossRef]
- Mair, C.M.; Ludwig, K.; Herrmann, A.; Sieben, C. Receptor binding and pH stability-How influenza A virus hemagglutinin affects host-specific virus infection. Biochim. Et Biophys. Acta (BBA)-Biomembr. 2014, 1838, 1153–1168. [Google Scholar] [CrossRef]
- Jeong, S.; Lee, D.H.; Kwon, J.H.; Kim, Y.J.; Lee, S.H.; Cho, A.Y.; Kim, T.H.; Park, J.E.; Lee, S.I.; Song, C.S. Highly Pathogenic Avian Influenza Clade 2.3.4.4b Subtype H5N8 Virus Isolated from Mandarin Duck in South Korea, 2020. Viruses 2020, 12, 1389. [Google Scholar] [CrossRef] [PubMed]
- Kandeil, A.; Moatasim, Y.; El Taweel, A.; El Sayes, M.; Rubrum, A.; Jeevan, T.; McKenzie, P.P.; Webby, R.J.; Ali, M.A.; Kayali, G.; et al. Genetic and Antigenic Characteristics of Highly Pathogenic Avian Influenza A(H5N8) Viruses Circulating in Domestic Poultry in Egypt, 2017-2021. Microorganisms 2022, 10, 595. [Google Scholar] [CrossRef] [PubMed]
- Salaheldin, A.H.; Elbestawy, A.R.; Abdelkader, A.M.; Sultan, H.A.; Ibrahim, A.A.; Abd El-Hamid, H.S.; Abdelwhab, E.M. Isolation of Genetically Diverse H5N8 Avian Influenza Viruses in Poultry in Egypt, 2019–2021. Viruses 2022, 14, 1431. [Google Scholar] [CrossRef]
- Kandeil, A.; Kayed, A.; Moatasim, Y.; Webby, R.J.; McKenzie, P.P.; Kayali, G.; Ali, M.A. Genetic characterization of highly pathogenic avian influenza A H5N8 viruses isolated from wild birds in Egypt. J. Gen. Virol 2017, 98, 1573–1586. [Google Scholar] [CrossRef] [PubMed]
- Chong, Y.; Ikematsu, H. Is seasonal vaccination a contributing factor to the selection of influenza epidemic variants? Hum. Vaccin. Immunother. 2018, 14, 518–522. [Google Scholar] [CrossRef]
- Duvvuri, V.R.; Duvvuri, B.; Cuff, W.R.; Wu, G.E.; Wu, J. Role of positive selection pressure on the evolution of H5N1 hemagglutinin. Genom. Proteom. Bioinform. 2009, 7, 47–56. [Google Scholar] [CrossRef]
- Li, W.; Shi, W.; Qiao, H.; Ho, S.Y.; Luo, A.; Zhang, Y.; Zhu, C. Positive selection on hemagglutinin and neuraminidase genes of H1N1 influenza viruses. Virol. J. 2011, 8, 183. [Google Scholar] [CrossRef]
- Zeng, X.-Y.; He, X.-W.; Meng, F.; Ma, Q.; Wang, Y.; Bao, H.-M.; Liu, Y.-J.; Deng, G.-H.; Shi, J.-Z.; Li, Y.-B.; et al. Protective efficacy of an H5/H7 trivalent inactivated vaccine (H5-Re13, H5-Re14, and H7-Re4 strains) in chickens, ducks, and geese against newly detected H5N1, H5N6, H5N8, and H7N9 viruses. J. Integr. Agric. 2022, 21, 2086–2094. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HA # | NA Stalk Del | PB2 | PB1-F2 Length | M “Amantadine Resistance Markers” | NS1 Length | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Receptor Binding Sites | Cleavage Site | 627 | 701 | ||||||||||||||
| 103 | 129 | 186 | 221 | 222 | 224 | 26 | 27 | 30 | 31 | 34 | |||||||
| Duck/Jiangsu/k1203/2010 | H | L | E | G | Q | G | PLREKRRKRGLF | No | E | D | 90 | L | V | A | N | G | 225 |
| A/duck/Zhejiang/6D18/2013 | H | L | E | G | Q | G | PLREKRRKRGLF | No | E | D | 52 | L | V | A | N | G | 230 |
| A/turkey/Germany/AR3390-L00939/2014 | H | L | E | G | Q | G | PLRERRRKRGLF | No | E | D | 52 | L | V | A | N | G | 237 |
| A/great_crested_grebe/Uvs-Nuur_Lake/341/2016 | H | L | E | G | Q | G | PLREKRRKRGLF | No | E | D | 52 | L | V | A | S | G | 230 |
| A/chicken/Iraq/1/2020 | H | L | E | G | Q | G | PLREKRRKRGLF | No | E | D | 52 | L | V | A | S | G | 217 |
| A/barnacle_goose/Netherlands/20017557-002/2020 | H | L | E | G | Q | G | PLREKRRKRGLF | No | E | D | 52 | L | V | A | S | G | 217 |
| A/domestic_duck/Germany-NI/AI03099/2021 | H | L | E | G | Q | G | PLKEKRRKRGLF | No | E | D | 52 | L | V | A | S | G | 217 |
| A/common-coot/Egypt/CA285/20165 | H | L | E | G | Q | G | PLREKRRKRGLF | No | E | D | 52 | L | V | A | S | G | 217 |
| A/chicken/Egypt/AF14/2020 * | H | L | E | G | Q | G | PLREKRRKRGLF | No | E | D | 52 | L | V | A | S | G | 230 |
| A/chicken/Egypt/AF12/2020 * | H | L | E | G | Q | G | PLREKRRKRGLF | No | E | D | 52 | L | V | A | S | G | 230 |
| A/chicken/Egypt/FAOS18E/2020 * | H | F | E | G | Q | G | PLREKRRKRGLF | No | E | D | 52 | L | V | A | S | G | 230 |
| A/turkey/Egypt/FAOSG6/2020 * | H | L | E | G | Q | G | PLREKRRKRGLF | No | E | D | 52 | L | V | A | S | G | 230 |
| Gene | a.a Residue | MEME | FEL | ||
|---|---|---|---|---|---|
| (p-Value ≤ 0.05) | Number of Branches under Episodic Selection | (p-Value ≤ 0.05) | LRT | ||
| HA gene | 23 | 0.05 | 1 | 0.04 | 4.5 |
| 88 | 0.04 | 2 | 0.03 | 4.9 | |
| 94 | 0.04 | 9 | 0.03 | 4.8 | |
| 175 | 0.01 | 7 | |||
| 195 | 0.02 | 4 | |||
| 438 | 0 | 1 | |||
| NA gene | 8 | 0.02 | 8 | 0.011 | 6.471 |
| 19 | 0.04 | 1 | |||
| 29 | 0.05 | 6 | 0.0347 | 4.462 | |
| 37 | 0.04 | 6 | 0.0204 | 5.374 | |
| 38 | 0.02 | 5 | 0.0118 | 6.335 | |
| 65 | 0.03 | 0 | |||
| 66 | 0.0358 | 4.408 | |||
| 71 | 0.0323 | 4.584 | |||
| 79 | 0.02 | 2 | |||
| 80 | 0.01 | 7 | 0.0101 | 6.62 | |
| 89 | 0.0429 | 4.098 | |||
| 151 | 0.03 | 5 | 0.0394 | 4.244 | |
| 177 | 0.0232 | 5.153 | |||
| 213 | 0.045 | 4.019 | |||
| 244 | 0.02 | 6 | 0.0191 | 5.492 | |
| 245 | 0.05 | 7 | 0.0402 | 4.211 | |
| 254 | 0.03 | 7 | 0.0339 | 4.501 | |
| 269 | 0.04 | 4 | 0.0453 | 4.008 | |
| 270 | 0.0484 | 3.896 | |||
| 300 | 0.01 | 7 | 0.0026 | 9.046 | |
| 311 | 0.02 | 1 | |||
| 314 | 0.0444 | 4.041 | |||
| 350 | 0.0392 | 4.253 | |||
| 382 | 0.02 | 7 | 0.0049 | 7.917 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hagag, N.M.; Yehia, N.; El-Husseiny, M.H.; Adel, A.; Shalaby, A.G.; Rabie, N.; Samy, M.; Mohamed, M.; El-Oksh, A.S.A.; Selim, A.; et al. Molecular Epidemiology and Evolutionary Analysis of Avian Influenza A(H5) Viruses Circulating in Egypt, 2019–2021. Viruses 2022, 14, 1758. https://doi.org/10.3390/v14081758
Hagag NM, Yehia N, El-Husseiny MH, Adel A, Shalaby AG, Rabie N, Samy M, Mohamed M, El-Oksh ASA, Selim A, et al. Molecular Epidemiology and Evolutionary Analysis of Avian Influenza A(H5) Viruses Circulating in Egypt, 2019–2021. Viruses. 2022; 14(8):1758. https://doi.org/10.3390/v14081758
Chicago/Turabian StyleHagag, Naglaa M., Nahed Yehia, Mohamed H. El-Husseiny, Amany Adel, Azhar G. Shalaby, Neveen Rabie, Mohamed Samy, Motaz Mohamed, Amal S. A. El-Oksh, Abdullah Selim, and et al. 2022. "Molecular Epidemiology and Evolutionary Analysis of Avian Influenza A(H5) Viruses Circulating in Egypt, 2019–2021" Viruses 14, no. 8: 1758. https://doi.org/10.3390/v14081758
APA StyleHagag, N. M., Yehia, N., El-Husseiny, M. H., Adel, A., Shalaby, A. G., Rabie, N., Samy, M., Mohamed, M., El-Oksh, A. S. A., Selim, A., Arafa, A.-S., Eid, S., Shahein, M. A., & Naguib, M. M. (2022). Molecular Epidemiology and Evolutionary Analysis of Avian Influenza A(H5) Viruses Circulating in Egypt, 2019–2021. Viruses, 14(8), 1758. https://doi.org/10.3390/v14081758