
CIGB-300 Peptide Targets the CK2 Phospho-Acceptor Domain on Human Papillomavirus E7 and Disrupts the Retinoblastoma (RB) Complex in Cervical Cancer Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Compounds
2.3. Plasmid Constructs
2.4. Cell Transient Transfection
2.5. Cell Viability Assay and Drug Treatments
2.6. Production and Purification of GST-Fusion Proteins
2.7. In Vitro Binding Assay Using GST-Fusion Proteins
2.8. In Vitro/In Vivo Pull-Down Assay
2.9. In Vitro/In Vivo Phosphorylation Assay
2.10. Immunoprecipitation Assay
2.11. Proteins Detection by Western Blot and Antibodies
2.12. Statistical Analysis
3. Results
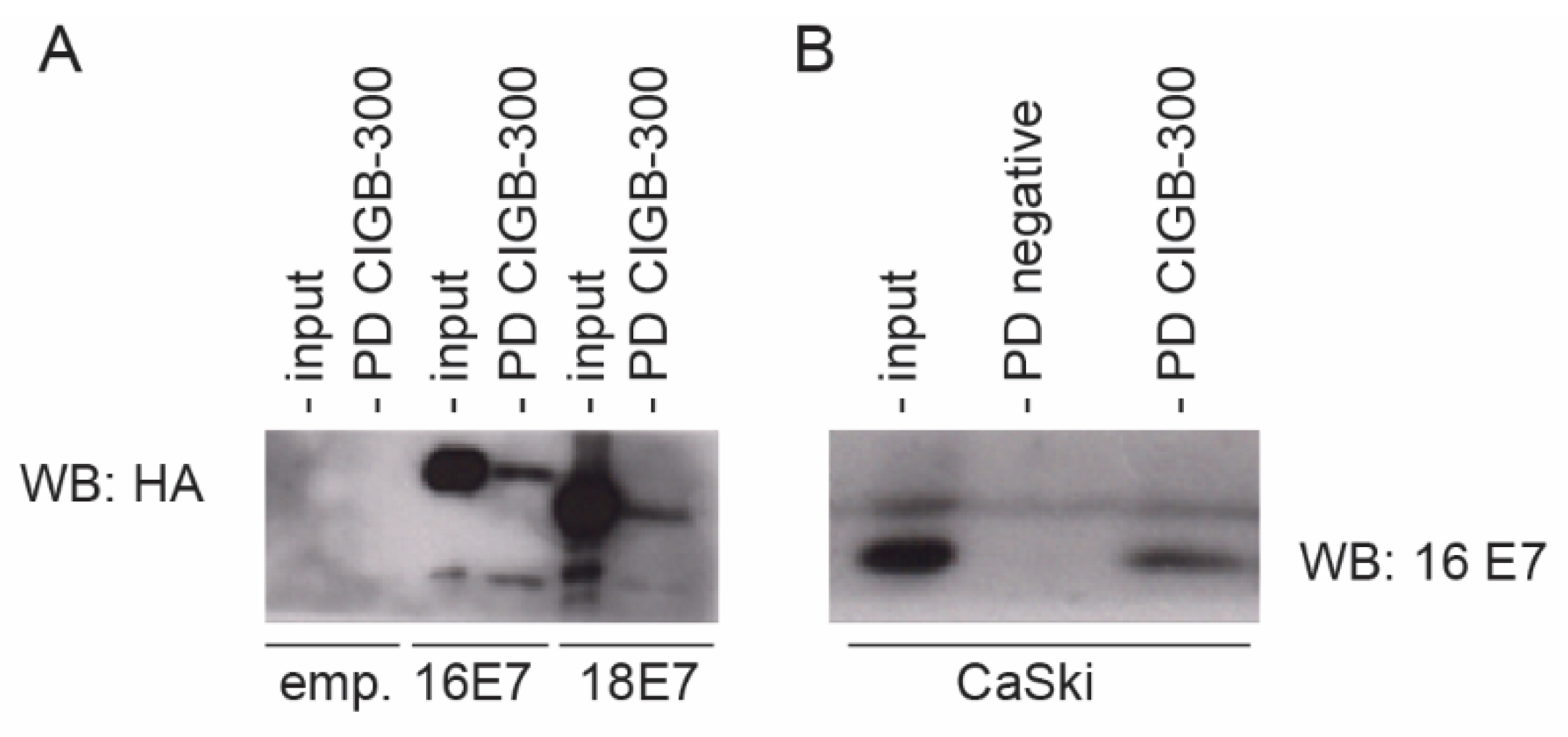
3.1. CIGB-300 Interacts with E7 Protein In Vitro
3.2. CIGB-300 Interacts with E7 Protein In Vivo
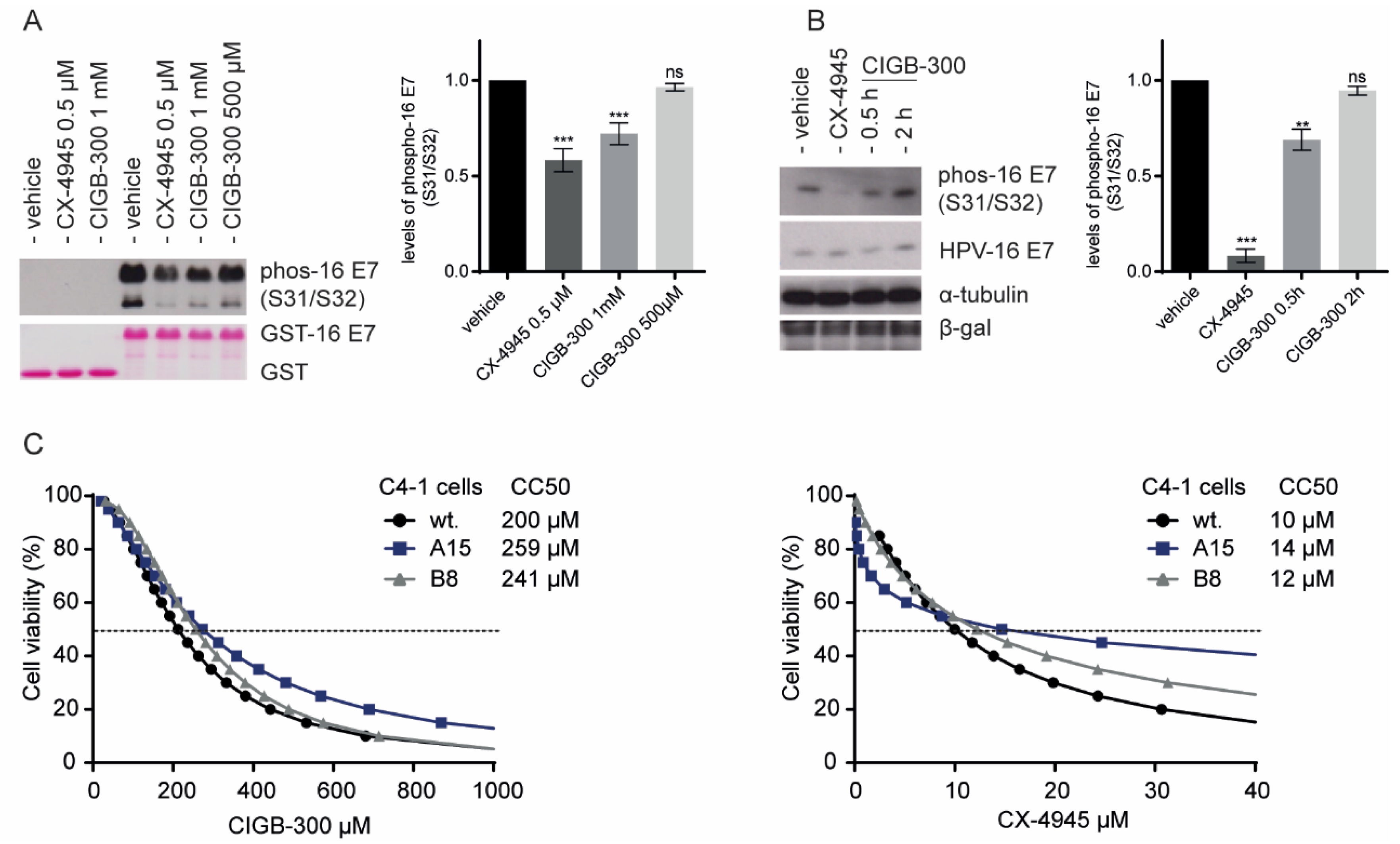
3.3. Inhibition of E7 Phosphorylation Is Not Essential for CIGB-300 Cytotoxicity in Cervical Cancer Cells
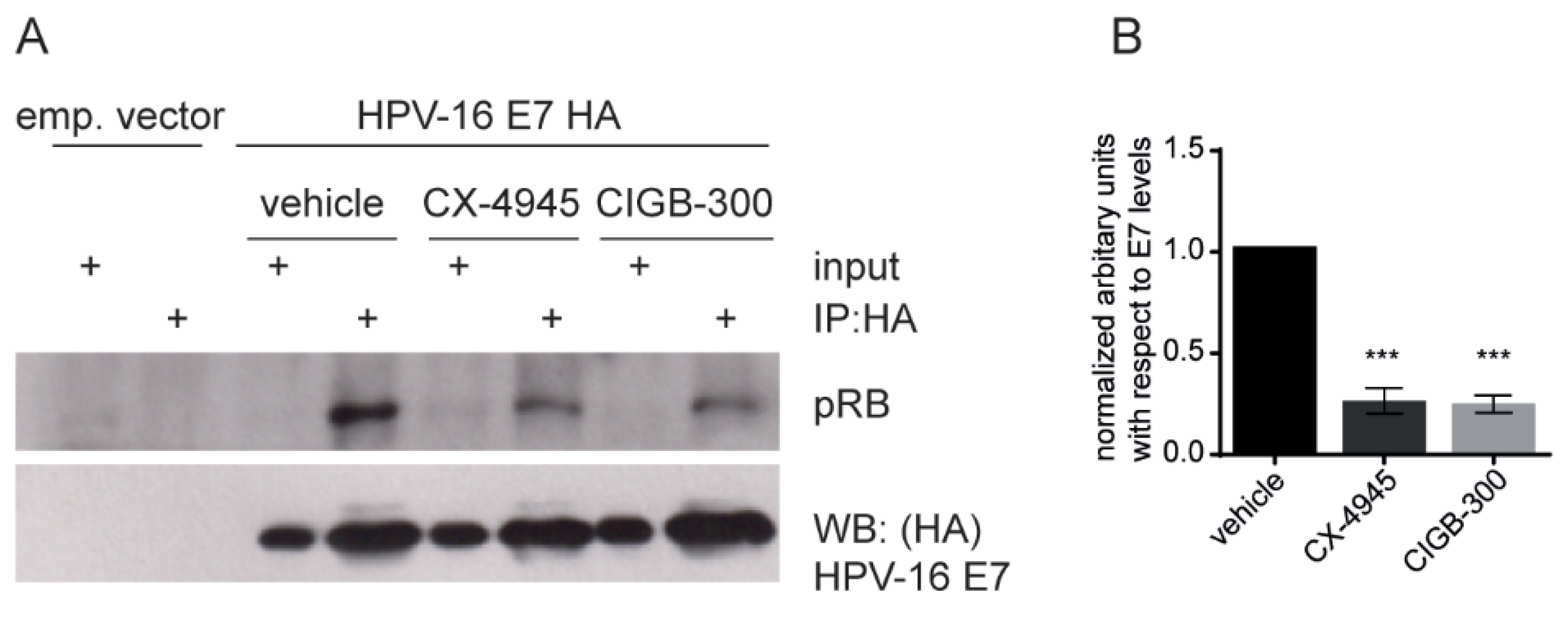
3.4. CIGB-300 Affects the HPV-16 E7–pRB Complex Formation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Egawa, N.; Egawa, K.; Griffin, H.; Doorbar, J. Human Papillomaviruses; Epithelial Tropisms, and the Development of Neoplasia. Viruses 2015, 7, 3863–3890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kombe Kombe, A.J.; Li, B.; Zahid, A.; Mengist, H.M.; Bounda, G.A.; Zhou, Y.; Jin, T. Epidemiology and Burden of Human Papillomavirus and Related Diseases, Molecular Pathogenesis, and Vaccine Evaluation. Front. Public Health 2020, 8, 552028. [Google Scholar] [CrossRef]
- Arbyn, M.; Weiderpass, E.; Bruni, L.; de Sanjosé, S.; Saraiya, M.; Ferlay, J.; Bray, F. Estimates of incidence and mortality of cervical cancer in 2018: A worldwide analysis. Lancet. Glob. Health 2020, 8, e191–e203. [Google Scholar] [CrossRef] [Green Version]
- Waheed, D.E.; Schiller, J.; Stanley, M.; Franco, E.L.; Poljak, M.; Kjaer, S.K.; Del Pino, M.; van der Klis, F.; Schim van der Loeff, M.F.; Baay, M.; et al. Human papillomavirus vaccination in adults: Impact, opportunities and challenges—A meeting report. BMC Proc. 2021, 15 (Suppl. 7), 16. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, M.; Li, X.; Yin, S.; Wang, B. An Overview of Novel Agents for Cervical Cancer Treatment by Inducing Apoptosis: Emerging Drugs Ongoing Clinical Trials and Preclinical Studies. Front. Med. 2021, 8, 682366. [Google Scholar] [CrossRef]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The biology and life-cycle of human papillomaviruses. Vaccine 2012, 30 (Suppl. 5), F55–F70. [Google Scholar] [CrossRef]
- Morgan, I.M.; DiNardo, L.J.; Windle, B. Integration of Human Papillomavirus Genomes in Head and Neck Cancer: Is It Time to Consider a Paradigm Shift? Viruses 2017, 9, 208. [Google Scholar] [CrossRef]
- Roman, A.; Munger, K. The papillomavirus E7 proteins. Virology 2013, 445, 138–168. [Google Scholar] [CrossRef] [Green Version]
- Vande Pol, S.B.; Klingelhutz, A.J. Papillomavirus E6 oncoproteins. Virology 2013, 445, 115–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huh, K.; Zhou, X.; Hayakawa, H.; Cho, J.Y.; Libermann, T.A.; Jin, J.; Harper, J.W.; Munger, K. Human papillomavirus type 16 E7 oncoprotein associates with the cullin 2 ubiquitin ligase complex, which contributes to degradation of the retinoblastoma tumor suppressor. J. Virol. 2007, 81, 9737–9747. [Google Scholar] [CrossRef] [Green Version]
- Poirson, J.; Biquand, E.; Straub, M.L.; Cassonnet, P.; Nominé, Y.; Jones, L.; van der Werf, S.; Travé, G.; Zanier, K.; Jacob, Y.; et al. Mapping the interactome of HPV E6 and E7 oncoproteins with the ubiquitin-proteasome system. FEBS J. 2017, 284, 3171–3201. [Google Scholar] [CrossRef] [Green Version]
- Firzlaff, J.M.; Lüscher, B.; Eisenman, R.N. Negative charge at the casein kinase II phosphorylation site is important for transformation but not for Rb protein binding by the E7 protein of human papillomavirus type 16. Proc. Natl. Acad. Sci. USA 1991, 88, 5187–5191. [Google Scholar] [CrossRef] [Green Version]
- Massimi, P.; Pim, D.; Storey, A.; Banks, L. HPV-16 E7 and adenovirus E1a complex formation with TATA box binding protein is enhanced by casein kinase II phosphorylation. Oncogene 1996, 12, 2325–2330. [Google Scholar] [PubMed]
- Basukala, O.; Mittal, S.; Massimi, P.; Bestagno, M.; Banks, L. The HPV-18 E7 CKII phospho acceptor site is required for maintaining the transformed phenotype of cervical tumour-derived cells. PLoS Pathog. 2019, 15, e1007769. [Google Scholar] [CrossRef] [PubMed]
- Perea, S.E.; Reyes, O.; Puchades, Y.; Mendoza, O.; Vispo, N.S.; Torrens, I.; Santos, A.; Silva, R.; Acevedo, B.; López, E.; et al. Antitumor effect of a novel proapoptotic peptide that impairs the phosphorylation by the protein kinase 2 (casein kinase 2). Cancer Res. 2004, 64, 7127–7129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perera, Y.; Ramos, Y.; Padrón, G.; Caballero, E.; Guirola, O.; Caligiuri, L.G.; Lorenzo, N.; Gottardo, F.; Farina, H.G.; Filhol, O.; et al. CIGB-300 anticancer peptide regulates the protein kinase CK2-dependent phosphoproteome. Mol. Cell. Biochem. 2020, 470, 63–75. [Google Scholar] [CrossRef]
- Rosales, M.; Pérez, G.V.; Ramón, A.C.; Cruz, Y.; Rodríguez-Ulloa, A.; Besada, V.; Ramos, Y.; Vázquez-Blomquist, D.; Caballero, E.; Aguilar, D.; et al. Targeting of Protein Kinase CK2 in Acute Myeloid Leukemia Cells Using the Clinical-Grade Synthetic-Peptide CIGB-300. Biomedicines 2021, 9, 766. [Google Scholar] [CrossRef]
- Perera, Y.; Farina, H.G.; Gil, J.; Rodriguez, A.; Benavent, F.; Castellanos, L.; Gómez, R.E.; Acevedo, B.E.; Alonso, D.F.; Perea, S.E. Anticancer peptide CIGB-300 binds to nucleophosmin/B23, impairs its CK2-mediated phosphorylation, and leads to apoptosis through its nucleolar disassembly activity. Mol. Cancer Ther. 2009, 8, 1189–1196. [Google Scholar] [CrossRef] [Green Version]
- Perera, Y.; Costales, H.C.; Diaz, Y.; Reyes, O.; Farina, H.G.; Mendez, L.; Gómez, R.E.; Acevedo, B.E.; Gomez, D.E.; Alonso, D.F.; et al. Sensitivity of tumor cells towards CIGB-300 anticancer peptide relies on its nucleolar localization. J. Pept. Sci. Off. Publ. Eur. Pept. Soc. 2012, 18, 215–223. [Google Scholar] [CrossRef]
- Águila, J.D.F.; Vega, Y.G.; Jiménez, R.O.R.; Sacerio, A.L.; Rodríguez, C.R.R.; Fraga, Y.R.; Silva, C.V. Safety of intravenous application of cigb-300 in patients with hematological malignancies. EHPMA study. Rev. Cuba. Hematol. Inmunol. Hemoter. 2016, 32, 236–248. [Google Scholar]
- García-Diegues, R.; de la Torre-Santos, A. Phase I Study of CIGB-300 Administered Intravenously in Patients with Relapsed/Refractory Solid Tumors. Arch. Med. 2018, 1, 4. [Google Scholar]
- Cruz, L.R.; Baladrón, I.; Rittoles, A.; Díaz, P.A.; Valenzuela, C.; Santana, R.; Vázquez, M.M.; García, A.; Chacón, D.; Thompson, D.; et al. Treatment with an Anti-CK2 Synthetic Peptide Improves Clinical Response in COVID-19 Patients with Pneumonia. A Randomized and Controlled Clinical Trial. ACS Pharmacol. Transl. Sci. 2021, 4, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Solares, A.M.; Santana, A.; Baladrón, I.; Valenzuela, C.; González, C.A.; Díaz, A.; Castillo, D.; Ramos, T.; Gómez, R.; Alonso, D.F.; et al. Safety and preliminary efficacy data of a novel casein kinase 2 (CK2) peptide inhibitor administered intralesionally at four dose levels in patients with cervical malignancies. BMC Cancer 2009, 9, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soriano-García, J.; López-Díaz, A.; Solares-Asteasuainzarra, M.; Baladrón-Castrillo, I.; Batista-Albuerne, N.; García-García, I.; González-Méndez, L.; Perera-Negrín, Y.; Valenzuela-Silva, C.; Pedro, A.J.J.C.R.T. Pharmacological and safety evaluation of CIGB-300, a casein kinase 2 inhibitor peptide, administered intralesionally to patients with cervical cancer stage IB2/II. J. Cancer Res. Ther. 2013, 1, 163–173. [Google Scholar]
- Sarduy, M.R.; García, I.; Coca, M.A.; Perera, A.; Torres, L.A.; Valenzuela, C.M.; Baladrón, I.; Solares, M.; Reyes, V.; Hernández, I.; et al. Optimizing CIGB-300 intralesional delivery in locally advanced cervical cancer. Br. J. Cancer 2015, 112, 1636–1643. [Google Scholar] [CrossRef] [Green Version]
- Perea, S.E.; Baladrón, I.; Valenzuela, C.; Perera, Y. CIGB-300: A peptide-based drug that impairs the Protein Kinase CK2-mediated phosphorylation. Semin. Oncol. 2018, 45, 58–67. [Google Scholar] [CrossRef]
- Perera, Y.; Melão, A.; Ramón, A.C.; Vázquez, D.; Ribeiro, D.; Perea, S.E.; Barata, J.T. Clinical-Grade Peptide-Based Inhibition of CK2 Blocks Viability and Proliferation of T-ALL Cells and Counteracts IL-7 Stimulation and Stromal Support. Cancers 2020, 12, 1377. [Google Scholar] [CrossRef]
- Pim, D.; Massimi, P.; Dilworth, S.M.; Banks, L. Activation of the protein kinase B pathway by the HPV-16 E7 oncoprotein occurs through a mechanism involving interaction with PP2A. Oncogene 2005, 24, 7830–7838. [Google Scholar] [CrossRef] [Green Version]
- Szalmás, A.; Tomaić, V.; Basukala, O.; Massimi, P.; Mittal, S.; Kónya, J.; Banks, L. The PTPN14 Tumor Suppressor Is a Degradation Target of Human Papillomavirus E7. J. Virol. 2017, 91, 7. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, S.L.; Stremlau, M.; He, X.; Basile, J.R.; Münger, K. Degradation of the retinoblastoma tumor suppressor by the human papillomavirus type 16 E7 oncoprotein is important for functional inactivation and is separable from proteasomal degradation of E7. J. Virol. 2001, 75, 7583–7591. [Google Scholar] [CrossRef] [Green Version]
- Vats, A. Understanding the Biology of HPV E6 Oncoprotein in Cervical Cancer. Doctoral Thesis, The Open University, Milton Keynes, UK, 2021. [Google Scholar]
- Keating, J.A.; Striker, R. Phosphorylation events during viral infections provide potential therapeutic targets. Rev. Med. Virol. 2012, 22, 166–181. [Google Scholar] [CrossRef] [PubMed]
- Borgo, C.; D’Amore, C.; Sarno, S.; Salvi, M.; Ruzzene, M. Protein kinase CK2: A potential therapeutic target for diverse human diseases. Signal Transduct. Target. Ther. 2021, 6, 183. [Google Scholar] [CrossRef] [PubMed]
- Gapany, M.; Faust, R.A.; Tawfic, S.; Davis, A.; Adams, G.L.; Ahmed, K. Association of elevated protein kinase CK2 activity with aggressive behavior of squamous cell carcinoma of the head and neck. Mol. Med. 1995, 1, 659–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faust, R.A.; Gapany, M.; Tristani, P.; Davis, A.; Adams, G.L.; Ahmed, K. Elevated protein kinase CK2 activity in chromatin of head and neck tumors: Association with malignant transformation. Cancer Lett. 1996, 101, 31–35. [Google Scholar] [CrossRef]
- Chua, M.M.J.; Lee, M.; Dominguez, I. Cancer-Type dependent expression of CK2 transcripts. PLoS ONE 2017, 12, e0188854. [Google Scholar] [CrossRef] [Green Version]
- Piirsoo, A.; Piirsoo, M.; Kala, M.; Sankovski, E.; Lototskaja, E.; Levin, V.; Salvi, M.; Ustav, M. Activity of CK2α protein kinase is required for efficient replication of some HPV types. PLoS Pathog. 2019, 15, e1007788. [Google Scholar] [CrossRef]
- Perera, Y.; Toro, N.D.; Gorovaya, L.; Fernandez, D.E.C.J.; Farina, H.G.; Perea, S.E. Synergistic interactions of the anti-casein kinase 2 CIGB-300 peptide and chemotherapeutic agents in lung and cervical preclinical cancer models. Mol. Clin. Oncol. 2014, 2, 935–944. [Google Scholar] [CrossRef] [Green Version]
- Trembley, J.H.; Li, B.; Kren, B.T.; Gravely, A.A.; Caicedo-Granados, E.; Klein, M.A.; Ahmed, K. CX-4945 and siRNA-Mediated Knockdown of CK2 Improves Cisplatin Response in HPV(+) and HPV(−) HNSCC Cell Lines. Biomedicines 2021, 9, 571. [Google Scholar] [CrossRef]
- Na Rangsee, N.; Yanatatsaneejit, P.; Pisitkun, T.; Somparn, P.; Jintaridth, P.; Topanurak, S. Host proteome linked to HPV E7-mediated specific gene hypermethylation in cancer pathways. Infect. Agents Cancer 2020, 15, 7. [Google Scholar] [CrossRef]
- Perea, S.E.; Reyes, O.; Baladron, I.; Perera, Y.; Farina, H.; Gil, J.; Rodriguez, A.; Bacardi, D.; Marcelo, J.L.; Cosme, K.; et al. CIGB-300, a novel proapoptotic peptide that impairs the CK2 phosphorylation and exhibits anticancer properties both in vitro and in vivo. Mol. Cell. Biochem. 2008, 316, 163–167. [Google Scholar] [CrossRef]
- Massimi, P.; Banks, L. Differential phosphorylation of the HPV-16 E7 oncoprotein during the cell cycle. Virology 2000, 276, 388–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avvakumov, N.; Torchia, J.; Mymryk, J.S. Interaction of the HPV E7 proteins with the pCAF acetyltransferase. Oncogene 2003, 22, 3833–3841. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramón, A.C.; Basukala, O.; Massimi, P.; Thomas, M.; Perera, Y.; Banks, L.; Perea, S.E. CIGB-300 Peptide Targets the CK2 Phospho-Acceptor Domain on Human Papillomavirus E7 and Disrupts the Retinoblastoma (RB) Complex in Cervical Cancer Cells. Viruses 2022, 14, 1681. https://doi.org/10.3390/v14081681
Ramón AC, Basukala O, Massimi P, Thomas M, Perera Y, Banks L, Perea SE. CIGB-300 Peptide Targets the CK2 Phospho-Acceptor Domain on Human Papillomavirus E7 and Disrupts the Retinoblastoma (RB) Complex in Cervical Cancer Cells. Viruses. 2022; 14(8):1681. https://doi.org/10.3390/v14081681
Chicago/Turabian StyleRamón, Ailyn C., Om Basukala, Paola Massimi, Miranda Thomas, Yasser Perera, Lawrence. Banks, and Silvio E. Perea. 2022. "CIGB-300 Peptide Targets the CK2 Phospho-Acceptor Domain on Human Papillomavirus E7 and Disrupts the Retinoblastoma (RB) Complex in Cervical Cancer Cells" Viruses 14, no. 8: 1681. https://doi.org/10.3390/v14081681
APA StyleRamón, A. C., Basukala, O., Massimi, P., Thomas, M., Perera, Y., Banks, L., & Perea, S. E. (2022). CIGB-300 Peptide Targets the CK2 Phospho-Acceptor Domain on Human Papillomavirus E7 and Disrupts the Retinoblastoma (RB) Complex in Cervical Cancer Cells. Viruses, 14(8), 1681. https://doi.org/10.3390/v14081681