
Detection of HIV-1 Transmission Clusters from Dried Blood Spots within a Universal Test-and-Treat Trial in East Africa
 , , , ,
, , , ,  , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
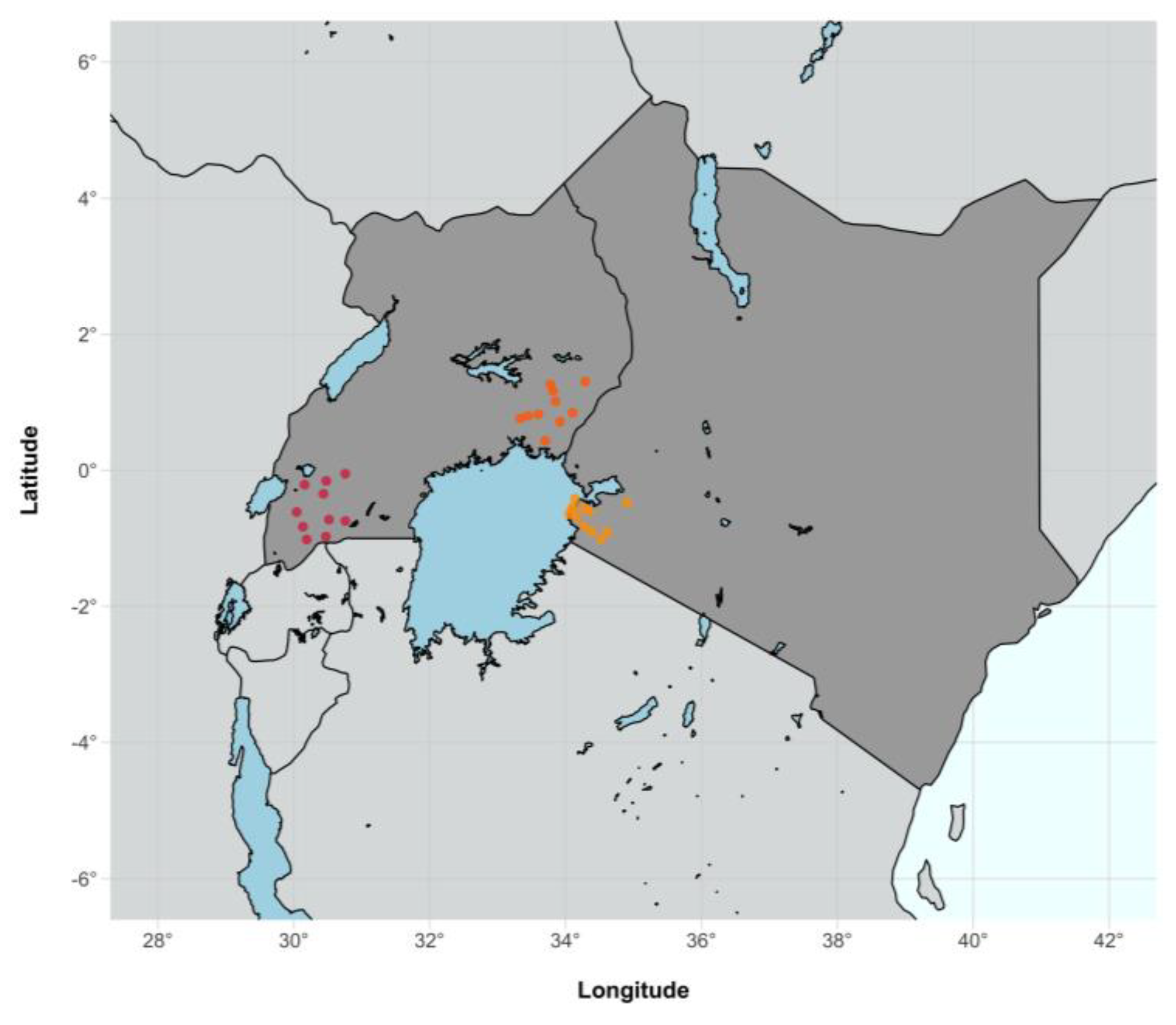
2.1. Study Design and Population
2.2. Sequence Subtyping
2.3. Drug Resistance Profiling
2.4. Genetic Linkage Analysis
2.5. Phylodynamic Analysis
3. Results
3.1. Characteristics of Sequenced SEARCH Trial Participants
3.2. Sequencing Density
3.3. Subtype Distribution
3.4. Drug Resistance Profiles
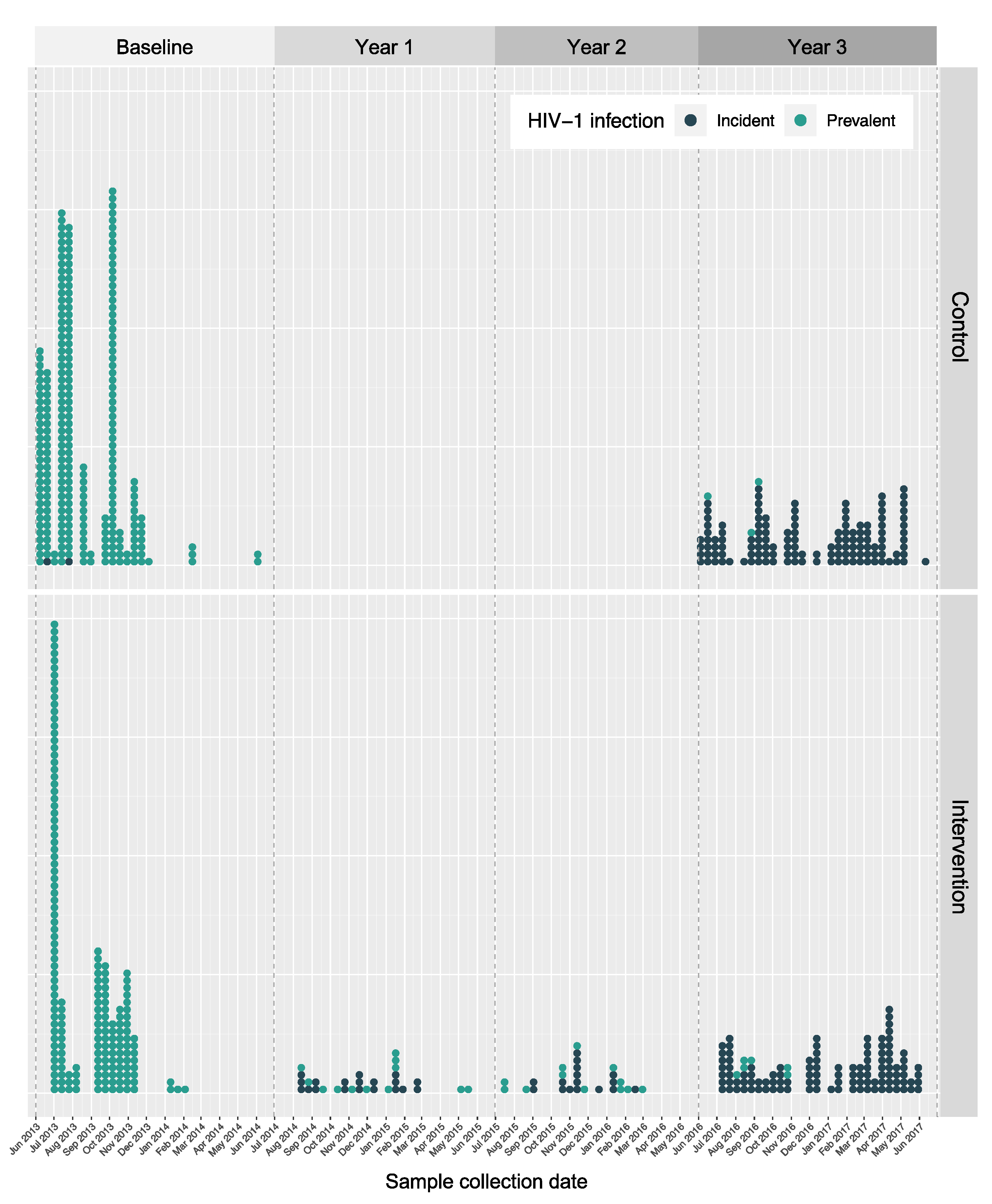
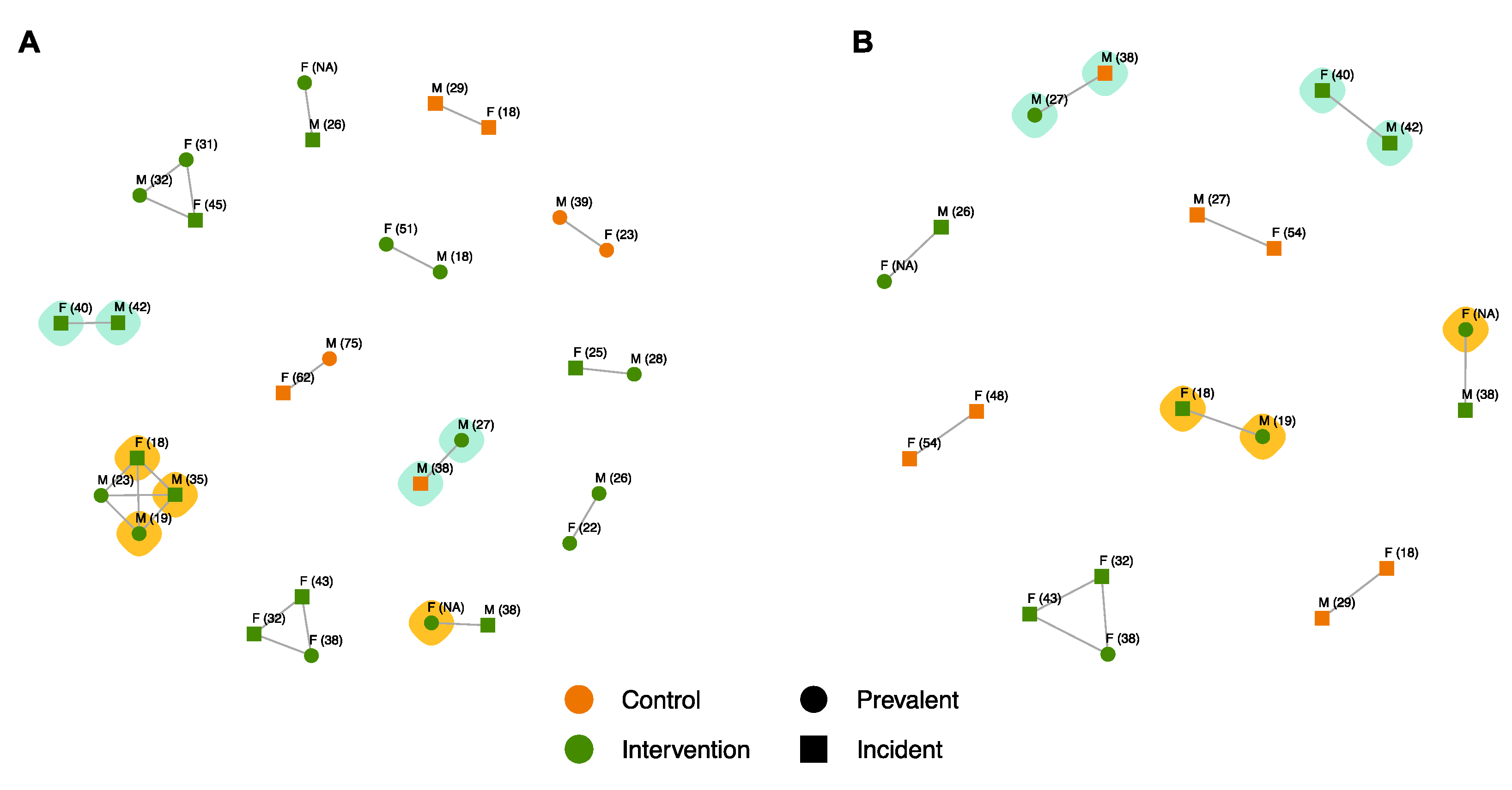
3.5. Identification of SEARCH Trial Clusters
3.5.1. Genetically Linked Clusters
3.5.2. SEARCH-Incident Transmission Events
3.6. Drug Resistance Characterisation of SEARCH Trial Clusters
4. Discussion
4.1. Regional Distribution of HIV-1 Subtypes
4.2. Drug Resistance in SEARCH
4.3. The Effect of the SEARCH Sampling Frequency on Genetic Linkage Clustering
4.4. Inferred SEARCH Trial Transmission Events
4.5. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Havlir, D.V.; Balzer, L.B.; Charlebois, E.D.; Clark, T.D.; Kwarisiima, D.; Ayieko, J.; Kabami, J.; Sang, N.; Liegler, T.; Chamie, G.; et al. HIV Testing and Treatment with the Use of a Community Health Approach in Rural Africa. N. Engl. J. Med. 2019, 381, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Hayes, R.J.; Donnell, D.; Floyd, S.; Mandla, N.; Bwalya, J.; Sabapathy, K.; Yang, B.; Phiri, M.; Schaap, A.; Eshleman, S.H.; et al. Effect of Universal Testing and Treatment on HIV Incidence—HPTN 071 (PopART). N. Engl. J. Med. 2019, 381, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Iwuji, C.C.; Orne-Gliemann, J.; Larmarange, J.; Balestre, E.; Thiebaut, R.; Tanser, F.; Okesola, N.; Makowa, T.; Dreyer, J.; Herbst, K.; et al. Universal Test and Treat and the HIV Epidemic in Rural South Africa: A Phase 4, Open-Label, Community Cluster Randomised Trial. Lancet HIV 2018, 5, e116–e125. [Google Scholar] [CrossRef]
- Makhema, J.; Wirth, K.E.; Pretorius Holme, M.; Gaolathe, T.; Mmalane, M.; Kadima, E.; Chakalisa, U.; Bennett, K.; Leidner, J.; Manyake, K.; et al. Universal Testing, Expanded Treatment, and Incidence of HIV Infection in Botswana. N. Engl. J. Med. 2019, 381, 230–242. [Google Scholar] [CrossRef]
- Havlir, D.; Lockman, S.; Ayles, H.; Larmarange, J.; Chamie, G.; Gaolathe, T.; Iwuji, C.; Fidler, S.; Kamya, M.; Floyd, S.; et al. What Do the Universal Test and Treat Trials Tell Us about the Path to HIV Epidemic Control? J. Int. AIDS Soc. 2020, 23, e25455. [Google Scholar] [CrossRef] [PubMed]
- Nduva, G.M.; Hassan, A.S.; Nazziwa, J.; Graham, S.M.; Esbjörnsson, J.; Sanders, E.J. HIV-1 Transmission Patterns Within and Between Risk Groups in Coastal Kenya. Sci. Rep. 2020, 10, 6775. [Google Scholar] [CrossRef] [PubMed]
- Yebra, G.; Ragonnet-Cronin, M.; Ssemwanga, D.; Parry, C.M.; Logue, C.H.; Cane, P.A.; Kaleebu, P.; Leigh Brown, A.J. Analysis of the History and Spread of HIV-1 in Uganda Using Phylodynamics. J. Gen. Virol. 2015, 96, 1890–1898. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, M.K.; Lessler, J.; Redd, A.D.; Kagaayi, J.; Laeyendecker, O.; Ndyanabo, A.; Nelson, M.I.; Cummings, D.A.T.; Bwanika, J.B.; Mueller, A.C.; et al. The Role of Viral Introductions in Sustaining Community-Based HIV Epidemics in Rural Uganda: Evidence from Spatial Clustering, Phylogenetics, and Egocentric Transmission Models. PLoS Med. 2014, 11, e1001610. [Google Scholar] [CrossRef]
- Bbosa, N.; Ssemwanga, D.; Nsubuga, R.N.; Salazar-Gonzalez, J.F.; Salazar, M.G.; Nanyonjo, M.; Kuteesa, M.; Seeley, J.; Kiwanuka, N.; Bagaya, B.S.; et al. Phylogeography of HIV-1 Suggests That Ugandan Fishing Communities Are a Sink for, Not a Source of, Virus from General Populations. Sci. Rep. 2019, 9, 1051. [Google Scholar] [CrossRef]
- Bbosa, N.; Ssemwanga, D.; Ssekagiri, A.; Xi, X.; Mayanja, Y.; Bahemuka, U.; Seeley, J.; Pillay, D.; Abeler-Dörner, L.; Golubchik, T.; et al. Phylogenetic and Demographic Characterization of Directed HIV-1 Transmission Using Deep Sequences from High-Risk and General Population Cohorts/Groups in Uganda. Viruses 2020, 12, 331. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, M.K.; Lessler, J.; Bazaale, J.; Nabukalu, D.; Nankinga, J.; Nantume, B.; Ssekasanvu, J.; Reynolds, S.J.; Ssekubugu, R.; Nalugoda, F.; et al. Migration, Hotspots, and Dispersal of HIV Infection in Rakai, Uganda. Nat. Commun. 2020, 11, 976. [Google Scholar] [CrossRef] [PubMed]
- Ratmann, O.; Kagaayi, J.; Hall, M.; Golubchick, T.; Kigozi, G.; Xi, X.; Wymant, C.; Nakigozi, G.; Abeler-Dörner, L.; Bonsall, D.; et al. Quantifying HIV Transmission Flow between High-Prevalence Hotspots and Surrounding Communities: A Population-Based Study in Rakai, Uganda. Lancet HIV 2020, 7, e173–e183. [Google Scholar] [CrossRef]
- Novitsky, V.; Zahralban-Steele, M.; Moyo, S.; Nkhisang, T.; Maruapula, D.; McLane, M.F.; Leidner, J.; Bennett, K.; Wirth, K.E.; Gaolathe, T.; et al. Mapping of HIV-1C Transmission Networks Reveals Extensive Spread of Viral Lineages across Villages in Botswana Treatment-as-Prevention Trial. J. Infect. Dis. 2020, 222, 1670–1680. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, J.L.; Burstein, G.R.; Pincus, J.; Branson, B. A Rapid Review of Rapid HIV Antibody Tests. Curr. Infect. Dis. Rep. 2006, 8, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Piwowar-Manning, E.; Fogel, J.; Laeyendecker, O.; Wolf, S.; Cummings, V.; Marzinke, M.; Clarke, W.; Breaud, A.; Wendel, S.; Wang, L.; et al. Failure to Identify HIV-Infected Individuals in a Clinical Trial Using a Single HIV Rapid Test for Screening. HIV Clin. Trials 2014, 15, 62–68. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Salazar-Gonzalez, J.F.; Salazar, M.G.; Tully, D.C.; Ogilvie, C.B.; Learn, G.H.; Allen, T.M.; Heath, S.L.; Goepfert, P.; Bar, K.J. Use of Dried Blood Spots to Elucidate Full-Length Transmitted/Founder HIV-1 Genomes. Pathog. Immun. 2016, 1, 129–153. [Google Scholar] [CrossRef]
- Pineda-Peña, A.C.; Faria, N.R.; Imbrechts, S.; Libin, P.; Abecasis, A.B.; Deforche, K.; Gómez-López, A.; Camacho, R.J.; De Oliveira, T.; Vandamme, A.M. Automated Subtyping of HIV-1 Genetic Sequences for Clinical and Surveillance Purposes: Performance Evaluation of the New REGA Version 3 and Seven Other Tools. Infect. Genet. Evol. 2013, 19, 337–348. [Google Scholar] [CrossRef]
- Struck, D.; Lawyer, G.; Ternes, A.M.; Schmit, J.C.; Bercoff, D.P. COMET: Adaptive Context-Based Modeling for Ultrafast HIV-1 Subtype Identification. Nucleic Acids Res. 2014, 42, e144. [Google Scholar] [CrossRef]
- Kosakokvsky Pond, S.L.; Posada, D.; Stawiski, E.; Chappey, C.; Poon, A.F.Y.; Hughes, G.; Fearnhill, E.; Gravenor, M.B.; Leigh Brown, A.J.; Frost, S.D.W. An Evolutionary Model-Based Algorithm for Accurate Phylogenetic Breakpoint Mapping and Subtype Prediction in HIV-1. PLoS Comput. Biol. 2009, 5, e1000581. [Google Scholar] [CrossRef]
- Rhee, S.Y.; Gonzales, M.J.; Kantor, R.; Betts, B.J.; Ravela, J.; Shafer, R.W. Human Immunodeficiency Virus Reverse Transcriptase and Protease Sequence Database. Nucleic Acids Res. 2003, 31, 298–303. [Google Scholar] [CrossRef]
- World Health Organisation. HIV Drug Resistance Report; WHO: Geneva, Switzerland, 2019. [Google Scholar]
- World Health Organisation. HIV Drug Resistance Report; WHO: Geneva, Switzerland, 2021. [Google Scholar]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Ragonnet-Cronin, M.; Hodcroft, E.; Hué, S.; Fearnhill, E.; Delpech, V.; Leigh Brown, A.J.; Lycett, S. Automated Analysis of Phylogenetic Clusters. BMC Bioinform. 2013, 14, 317. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Weaver, S.; Leigh Brown, A.J.; Wertheim, J.O. HIV-TRACE (TRAnsmission Cluster Engine): A Tool for Large Scale Molecular Epidemiology of HIV-1 and Other Rapidly Evolving Pathogens. Mol. Biol. Evol. 2018, 35, 1812–1819. [Google Scholar] [CrossRef]
- Rose, R.; Lamers, S.L.; Dollar, J.J.; Grabowski, M.K.; Hodcroft, E.B.; Ragonnet-Cronin, M.; Wertheim, J.O.; Redd, A.D.; German, D.; Laeyendecker, O. Identifying Transmission Clusters with Cluster Picker and HIV-TRACE. AIDS Res. Hum. Retrovir. 2017, 33, 211–218. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian Phylogenetic and Phylodynamic Data Integration Using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef]
- Harris, M.E.; Serwadda, D.; Sewankambo, N.; Kim, B.; Kigozi, G.; Kiwanuka, N.; Phillips, J.B.; Wabwire, F.; Meehen, M.; Lutalo, T.; et al. Among 46 Near Full Length HIV Type 1 Genome Sequences from Rakai District, Uganda, Subtype D and AD Recombinants Predominate. AIDS Res. Hum. Retrovir. 2002, 18, 1281–1290. [Google Scholar] [CrossRef] [PubMed]
- Grant, H.E. Characterisation of the Ugandan HIV Epidemic with Full-Length Genome Sequence Data from 1986 to 2016. PhD Thesis, University of Edinburgh, Edinburgh, UK, 2022. [Google Scholar]
- Hill, V.; Baele, G. Bayesian Estimation of Past Population Dynamics in BEAST 1.10 Using the Skygrid Coalescent Model. Mol. Biol. Evol. 2019, 36, 2620–2628. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.T.; Carvalho, L.M.; Pybus, O.G. Exploring the Temporal Structure of Heterochronous Sequences Using TempEst (Formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef]
- Drummond, A.J.; Ho, S.Y.W.; Phillips, M.J.; Rambaut, A. Relaxed Phylogenetics and Dating with Confidence. PLoS Biol. 2006, 4, 699–710. [Google Scholar] [CrossRef]
- Hasegawa, M.; Kishino, H.; Yano, T. aki Dating of the Human-Ape Splitting by a Molecular Clock of Mitochondrial DNA. J. Mol. Evol. 1985, 22, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. Maximum Likelihood Phylogenetic Estimation from DNA Sequences with Variable Rates over Sites: Approximate Methods. J. Mol. Evol. 1994, 39, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Gill, M.S.; Lemey, P.; Faria, N.R.; Rambaut, A.; Shapiro, B.; Suchard, M.A. Improving Bayesian Population Dynamics Inference: A Coalescent-Based Model for Multiple Loci. Mol. Biol. Evol. 2013, 30, 713–724. [Google Scholar] [CrossRef]
- Minin, V.N.; Bloomquist, E.W.; Suchard, M.A. Smooth Skyride through a Rough Skyline: Bayesian Coalescent-Based Inference of Population Dynamics. Mol. Biol. Evol. 2008, 25, 1459–1471. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Nascimento, F.F.; dos Reis, M.; Yang, Z. A Biologist’s Guide to Bayesian Phylogenetic Analysis. Nat. Ecol. Evol. 2017, 1, 1446–1454. [Google Scholar] [CrossRef]
- Lihana, R.W.; Ssemwanga, D.; Abimiku, A.G.; Ndembi, N. Update on HIV-1 Diversity in Africa: A Decade in Review. AIDS Rev. 2012, 14, 83–100. [Google Scholar]
- Bbosa, N.; Kaleebu, P.; Ssemwanga, D. HIV Subtype Diversity Worldwide. Curr. Opin. HIV AIDS 2019, 14, 153–160. [Google Scholar] [CrossRef]
- Poon, A.F.Y.; Ndashimye, E.; Avino, M.; Gibson, R.; Kityo, C.; Kyeyune, F.; Nankya, I.; Quiñones-Mateu, M.E.; ARTS, E.J.; The Ugandan Drug Resistance Study Team. First-Line HIV Treatment Failures in Non-B Subtypes and Recombinants: A Cross-Sectional Analysis of Multiple Populations in Uganda. AIDS Res. Ther. 2019, 16, 3. [Google Scholar] [CrossRef]
- Hemelaar, J.; Elangovan, R.; Yun, J.; Dickson-Tetteh, L.; Kirtley, S.; Gouws-Williams, E.; Ghys, P.D.; Abimiku, A.G.; Agwale, S.; Archibald, C.; et al. Global and Regional Epidemiology of HIV-1 Recombinants in 1990–2015: A Systematic Review and Global Survey. Lancet HIV 2020, 7, e772–e781. [Google Scholar] [CrossRef]
- Ssemwanga, D.; Lihana, R.W.; Ugoji, C.; Abimiku, A.G.; Nkengasong, J.N.; Dakum, P.; Ndembi, N. Update on HIV-1 Acquired and Transmitted Drug Resistance in Africa. AIDS Rev. 2015, 17, 3–20. [Google Scholar] [PubMed]
- Poon, A.F.Y. Impacts and Shortcomings of Genetic Clustering Methods for Infectious Disease Outbreaks. Virus Evol. 2016, 2, vew031. [Google Scholar] [CrossRef] [PubMed]
- Camlin, C.S.; Akullian, A.; Neilands, T.B.; Getahun, M.; Bershteyn, A.; Ssali, S.; Geng, E.; Gandhi, M.; Cohen, C.R.; Maeri, I.; et al. Gendered Dimensions of Population Mobility Associated with HIV across Three Epidemics in Rural Eastern Africa. Health Place 2019, 57, 339–351. [Google Scholar] [CrossRef] [PubMed]
- Sigaloff, K.C.E.; Hamers, R.L.; Wallis, C.L.; Kityo, C.; Siwale, M.; Ive, P.; Botes, M.E.; Mandaliya, K.; Wellington, M.; Osibogun, A.; et al. Unnecessary Antiretroviral Treatment Switches and Accumulation of HIV Resistance Mutations; Two Arguments for Viral Load Monitoring in Africa. J. Acquir. Immune Defic. Syndr. 2011, 58, 23–31. [Google Scholar] [CrossRef] [PubMed]
- World Health Organisation. Consolidated Guidelines on HIV Prevention, Testing, Treatment, Service Delivery and Monitoring: Recommendations for a Public Health Approach; WHO: Geneva, Switzerland, 2021. [Google Scholar]
- Vitoria, M.; Hill, A.; Ford, N.; Doherty, M.; Clayden, P.; Venter, F.; Ripin, D.; Flexner, C.; Domanico, P.L. The Transition to Dolutegravir and Other New Antiretrovirals in Low-Income and Middle-Income Countries: What Are the Issues? AIDS 2018, 32, 1551–1561. [Google Scholar] [CrossRef]
- Ratmann, O.; Grabowski, M.K.; Hall, M.; Golubchik, T.; Wymant, C.; Abeler-Dörner, L.; Bonsall, D.; Hoppe, A.; Leigh Brown, A.J.; de Oliveira, T.; et al. Inferring HIV-1 Transmission Networks and Sources of Epidemic Spread in Africa with Deep-Sequence Phylogenetic Analysis. Nat. Commun. 2019, 10, 1411. [Google Scholar] [CrossRef] [PubMed]
- Tavaré, S. Some Probabilistic and Statistical Problems in the Analysis of DNA Sequences. Lect. Math. Life Sci. 1986, 17, 57–86. [Google Scholar]
- Kingman, J.F.C. The Coalescent. Stoch. Processes Appl. 1982, 13, 235–248. [Google Scholar] [CrossRef]
- Griffiths, R.C.; Tavaré, S. Sampling Theory for Neutral Alleles in a Varying Environment. Philos. Trans. R. Soc. London. Ser. B Biol. Sci. 1994, 344, 403–410. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Western Uganda | Eastern Uganda | Western Kenya | NA | Total | |
|---|---|---|---|---|---|
| n (%) | n (%) | n (%) | n (%) | n (%) | |
| All | 377 (50.6) | 61 (8.2) | 305 (40.9) | 2 (0.3) | 745 (100) |
| Gender | |||||
| (χ2 test p-value = 5 × 10−5) | |||||
| Female | 180 (47.7) | 31 (50.8) | 197 (64.6) | 2 (100) | 410 (55) |
| Male | 197 (52.3) | 30 (49.2) | 108 (35.4) | 0 | 335 (45) |
| Age category | |||||
| 15–20 years | 17 (4.5) | 4 (6.6) | 18 (5.9) | 0 | 39 (5.2) |
| 21–49 years | 311 (82.5) | 47 (77) | 236 (77.4) | 0 | 594 (79.7) |
| ≥50 years | 45 (11.9) | 10 (16.4) | 42 (13.8) | 0 | 97 (13) |
| NA | 4 (1.1) | 0 | 9 (3) | 2 (100) | 15 (2) |
| Occupation * | |||||
| (χ2 test p-value = 7 × 10−4) | |||||
| Formal sector | 50 (13.3) | 6 (9.8) | 66 (21.6) | 0 | 122 (16.4) |
| High-risk informal sector | 22 (5.8) | 4 (6.6) | 24 (7.9) | 0 | 50 (6.7) |
| Low-risk informal sector | 282 (74.8) | 51 (83.6) | 179 (58.7) | 0 | 512 (68.7) |
| Other | 12 (3.2) | 0 | 20 (6.6) | 0 | 32 (4.3) |
| No job or disabled | 7 (1.9) | 0 | 7 (2.3) | 0 | 14 (1.9) |
| NA | 4 (1.1) | 0 | 9 (3) | 2 (100) | 15 (2) |
| HIV-1 infection † | |||||
| (χ2 test p-value < 2 × 10−16) | |||||
| Prevalent | 263 (69.8) | 5 (8.2) | 199 (65.2) | 2 (100) | 469 (63) |
| Incident | 114 (30.2) | 56 (91.8) | 106 (34.8) | 0 | 276 (37) |
| Trial arm | |||||
| (χ2 test p-value = 6 × 10−9) | |||||
| Intervention | 142 (37.7) | 22 (36.1) | 184 (60.3) | 0 | 348 (46.7) |
| Control | 235 (62.3) | 39 (63.9) | 121 (39.7) | 0 | 395 (53) |
| NA | 0 | 0 | 0 | 2 (100) | 2 (0.3) |
| Total HIV+ | Sequenced HIV+ | Sequencing Density | |
|---|---|---|---|
| n | n | % | |
| Prevalent sequencing density by geographical region | |||
| Western Uganda | 2873 | 263 | 9.15 |
| Eastern Uganda | 1590 | 5 | 0.31 |
| Western Kenya | 9066 | 199 | 2.19 |
| Total | 13,529 | 467 | 3.45 |
| Incident sequencing density by geographical region | |||
| Western Uganda | 317 | 114 | 35.96 |
| Eastern Uganda | 132 | 56 | 42.42 |
| Western Kenya | 401 | 106 | 26.43 |
| Total | 850 | 276 | 32.47 |
| Prevalent sequencing density by trial arm | |||
| Intervention | 7212 | 204 | 2.83 |
| Control | 6317 | 263 | 4.16 |
| Total | 13,529 | 467 | 3.45 |
| Incident sequencing density by trial arm | |||
| Intervention | 435 | 144 | 33.10 |
| Control | 415 | 132 | 31.81 |
| Total | 850 | 276 | 32.47 |
| Western Uganda | Eastern Uganda | Western Kenya | Total | |||||
|---|---|---|---|---|---|---|---|---|
| n (%) | n (%) | n (%) | n (%) | |||||
| gag | pol | gag | pol | gag | pol | gag | pol | |
| A1 | 141 (49.1) | 122 (48.8) | 32 (68.1) | 35 (62.5) | 149 (69.6) | 119 (66.1) | 322 (58.8) | 276 (56.8) |
| A2 | 0 | 0 | 0 | 0 | 4 (1.9) | 3 (1.7) | 4 (0.7) | 3 (0.6) |
| D | 71 (24.7) | 61 (24.4) | 10 (21.3) | 14 (25) | 25 (11.7) | 21 (11.7) | 106 (19.3) | 96 (19.7) |
| C | 29 (10.1) | 20 (8) | 0 | 0 | 12 (5.6) | 8 (4.4) | 41 (7.5) | 28 (5.8) |
| G | 1 (0.4) | 1 (0.4) | 0 | 0 | 2 (0.9) | 0 | 3 (0.6) | 1 (0.2) |
| Recombinants | 45 (15.7) | 46 (18.4) | 5 (10.6) | 7 (12.5) | 22 (10.3) | 29 (16.1) | 72 (13.1) | 82 (16.9) |
| Total | 287 (100) | 250 (100) | 47 (100) | 56 (100) | 214 (100) | 180 (100) | 548 (100) | 486 (100) |
| Sensitive | NRTI Resistance Only | NNRTI Resistance Only | NRTI + NNRTI Resistance | Total | |
|---|---|---|---|---|---|
| n (%) | n (%) | n (%) | n (%) | n (%) | |
| Drug resistant sequences by trial arm | |||||
| Intervention | 203 (87.1) | 2 (0.9) | 27 (11.6) | 1 (0.4) | 233 (47.9) |
| Control | 214 (84.6) | 1 (0.4) | 34 (13.4) | 4 (1.6) | 253 (52.1) |
| Total | 417 (85.8) | 3 (0.6) | 61 (12.6) | 5 (1) | 486 (100) |
| Drug resistant sequences by HIV-1 infection † | |||||
| Prevalent | 208 (84.9) | 2 (0.8) | 31 (12.7) | 4 (1.6) | 245 (50.2) |
| Incident | 211 (86.8) | 1 (0.4) | 30 (12.3) | 1 (0.4) | 243 (49.8) |
| Total | 419 (85.9) | 3 (0.6) | 61 (12.5) | 5 (1) | 488 (100) |
| <1.5% GD Clusters | SEARCH-Incident Clusters | |
|---|---|---|
| n (%) | n (%) | |
| All | 13 (100) | 9 (100) |
| Gender | ||
| Differing gender cluster | 11 (84.6) | 6 (66.7) |
| Same gender (female) cluster | 1 (7.7) | 2 (22.2) |
| Same gender (male) cluster | 1 (7.7) | 1 (11.1) |
| HIV-1 infection category | ||
| Prevalent cluster | 3 (23.1) | 0 |
| Incident cluster | 2 (15.4) | 4 (44.4) |
| Mixed cluster | 8 (61.5) | 5 (55.6) |
| Region | ||
| Intra-region cluster | 13 (100) | 9 (100) |
| Inter-region cluster | 0 | 0 |
| Community | ||
| Intra-community cluster | 11 (84.6) | 7 (77.8) |
| Inter-community cluster | 2 (15.4) | 2 (22.2) |
| Age | ||
| Intra-age cluster | 8 (61.5) | 4 (44.4) |
| Inter-age cluster | 3 (23.1) | 3 (33.3) |
| NA | 2 (15.4) | 2 (22.2) |
| Occupation | ||
| Intra-occupation cluster | 4 (30.8) | 3 (33.3) |
| Inter-occupation cluster | 7 (53.8) | 4 (44.4) |
| NA | 2 (15.4) | 2 (22.2) |
| Trial arm | ||
| Intervention cluster | 9 (69.2) | 5 (55.6) |
| Control cluster | 3 (23.1) | 3 (33.3) |
| Mixed cluster | 1 (7.7) | 1 (11.1) |
| Subtype | ||
| A1 cluster | 10 (76.9) | 7 (77.8) |
| D cluster | 2 (15.4) | 2 (22.2) |
| Recombinant cluster | 1 (7.7) | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pujol-Hodge, E.; Salazar-Gonzalez, J.F.; Ssemwanga, D.; Charlebois, E.D.; Ayieko, J.; Grant, H.E.; Liegler, T.; Atkins, K.E.; Kaleebu, P.; Kamya, M.R.; et al. Detection of HIV-1 Transmission Clusters from Dried Blood Spots within a Universal Test-and-Treat Trial in East Africa. Viruses 2022, 14, 1673. https://doi.org/10.3390/v14081673
Pujol-Hodge E, Salazar-Gonzalez JF, Ssemwanga D, Charlebois ED, Ayieko J, Grant HE, Liegler T, Atkins KE, Kaleebu P, Kamya MR, et al. Detection of HIV-1 Transmission Clusters from Dried Blood Spots within a Universal Test-and-Treat Trial in East Africa. Viruses. 2022; 14(8):1673. https://doi.org/10.3390/v14081673
Chicago/Turabian StylePujol-Hodge, Emma, Jesus F. Salazar-Gonzalez, Deogratius Ssemwanga, Edwin D. Charlebois, James Ayieko, Heather E. Grant, Teri Liegler, Katherine E. Atkins, Pontiano Kaleebu, Moses R. Kamya, and et al. 2022. "Detection of HIV-1 Transmission Clusters from Dried Blood Spots within a Universal Test-and-Treat Trial in East Africa" Viruses 14, no. 8: 1673. https://doi.org/10.3390/v14081673
APA StylePujol-Hodge, E., Salazar-Gonzalez, J. F., Ssemwanga, D., Charlebois, E. D., Ayieko, J., Grant, H. E., Liegler, T., Atkins, K. E., Kaleebu, P., Kamya, M. R., Petersen, M., Havlir, D. V., & Leigh Brown, A. J. (2022). Detection of HIV-1 Transmission Clusters from Dried Blood Spots within a Universal Test-and-Treat Trial in East Africa. Viruses, 14(8), 1673. https://doi.org/10.3390/v14081673