
Robust Phylodynamic Analysis of Genetic Sequencing Data from Structured Populations

 , , , and
, , , and
Abstract
:1. Introduction
2. Methods
2.1. Description of the Extended Multi-Type Birth–Death Model
2.2. Implementation Improvements
3. Results
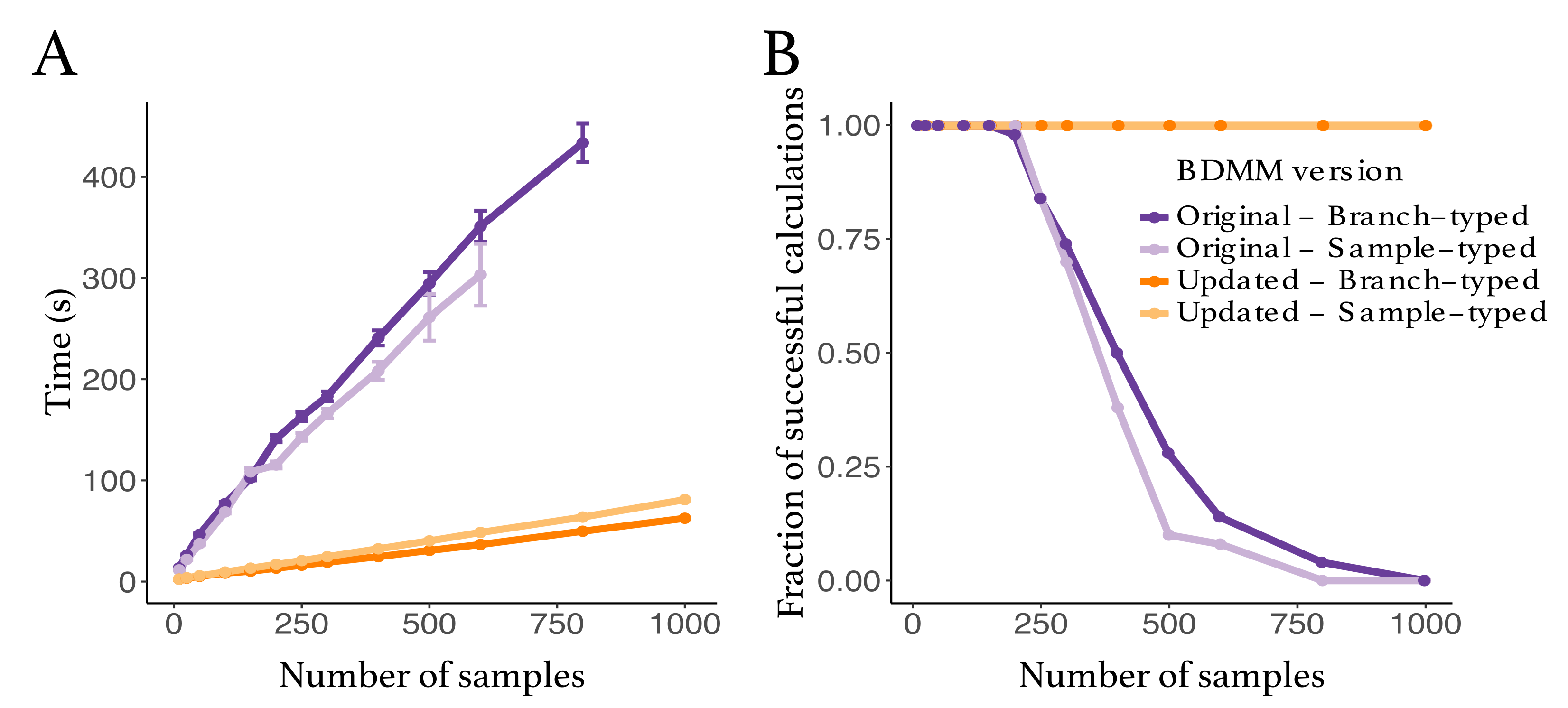
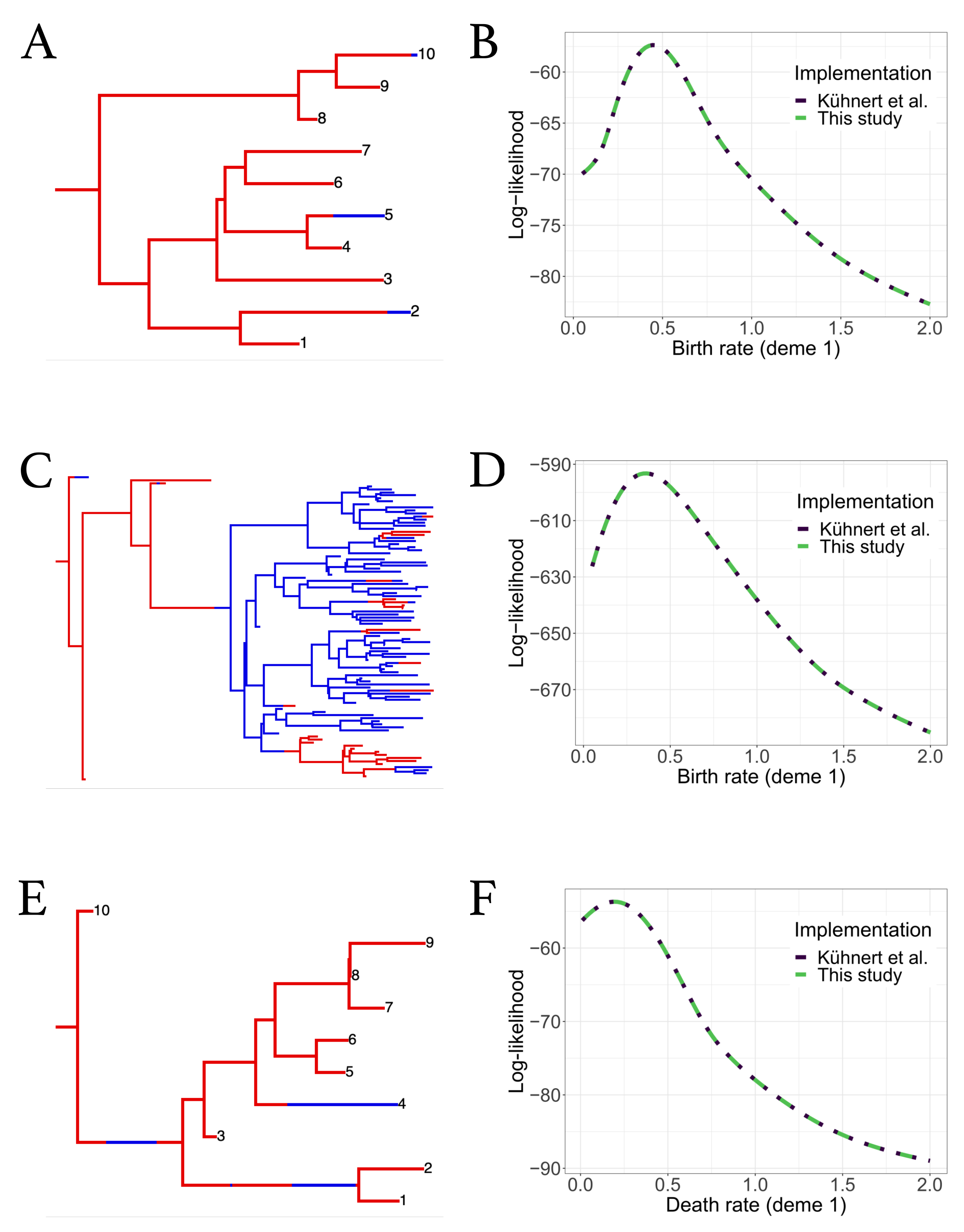
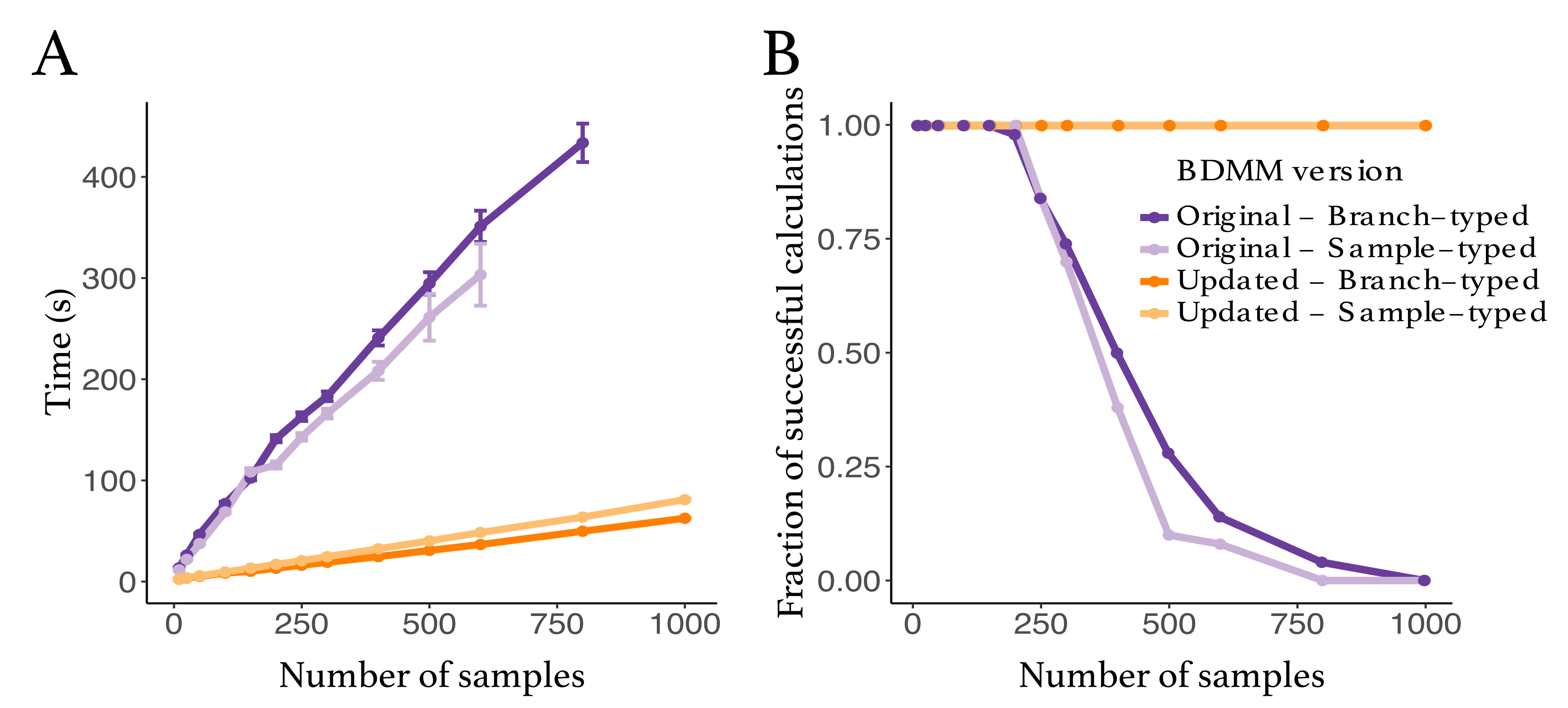
3.1. Evaluation of Numerical Improvements
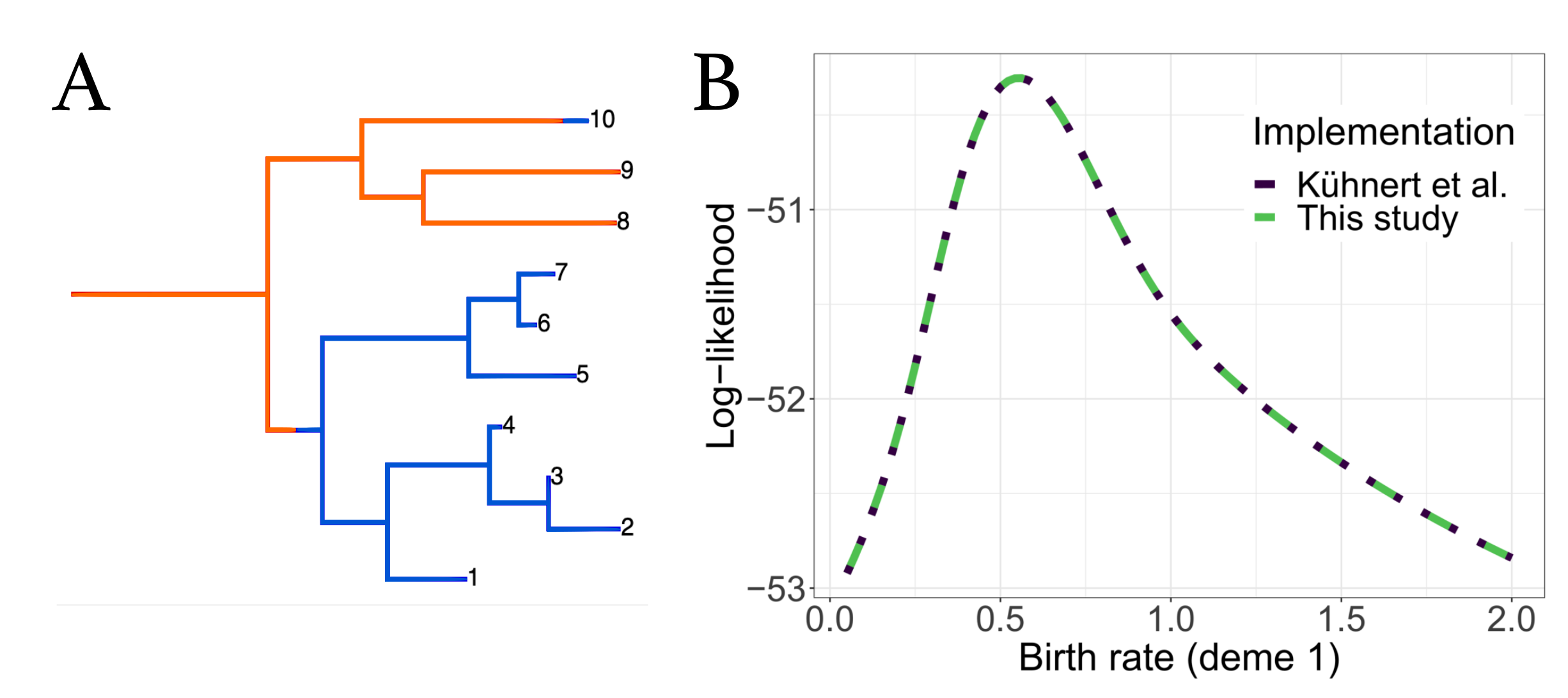
3.2. Validation against Original Implementation
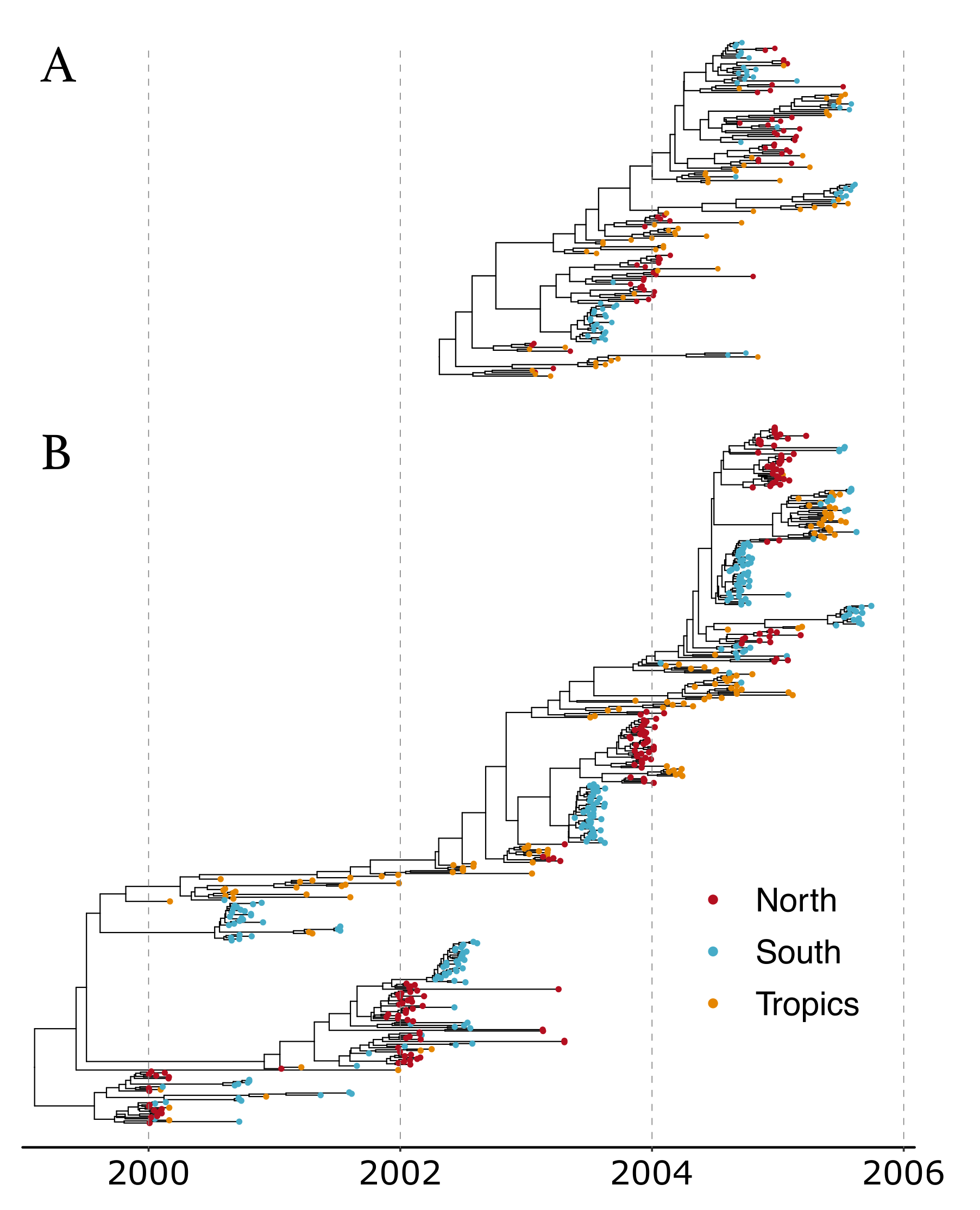
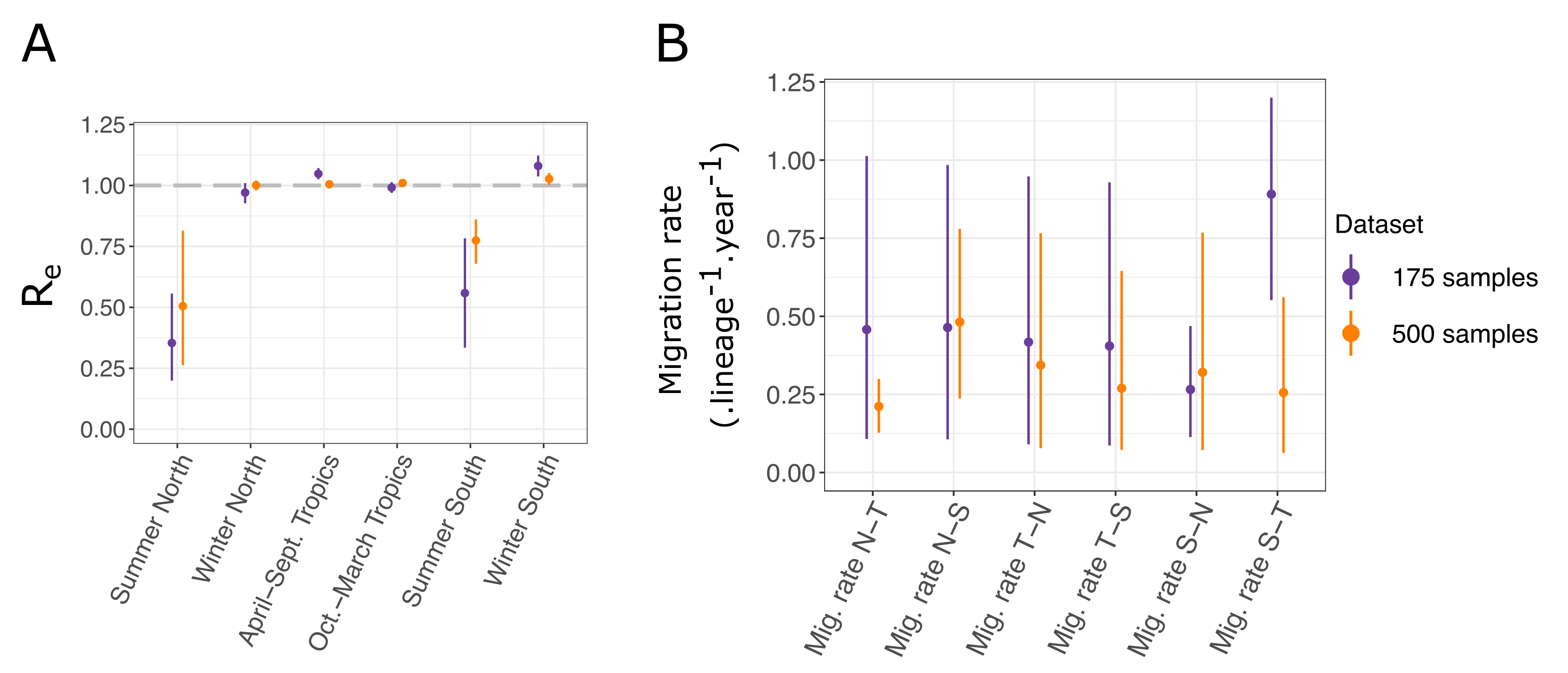
3.3. Influenza A Virus (H3N2) Analysis
3.4. Properties of bdmm
3.4.1. Identifiability of Parameters
3.4.2. Computational Costs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Derivation of the Probability Density of a Sampled Tree
Appendix A.1. Probability of Having No Sampled Descendants
Appendix A.2. Probability Density of a Sample-Typed Subtree
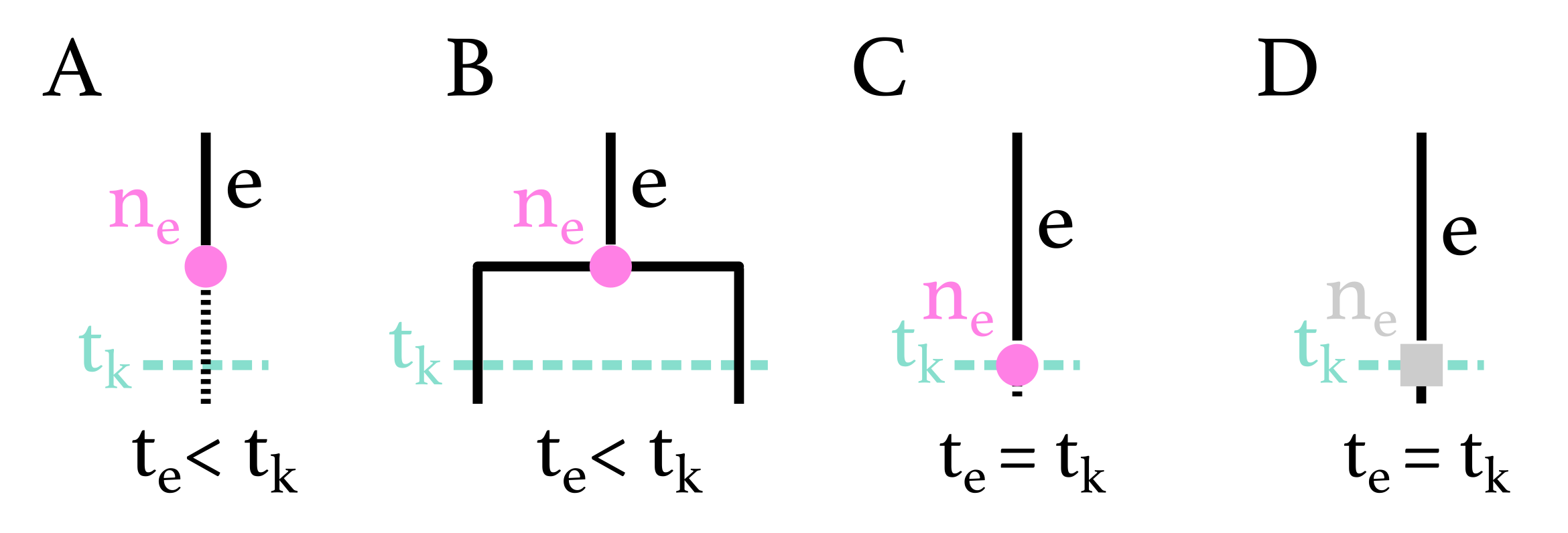
Appendix A.3. Probability Density of a Branch-Typed Subtree
Appendix A.4. Probability Density of a Sampled Tree
Appendix B. Improved Implementation of the Tree Probability Density Evaluation
Appendix B.1. Extended Numerical Representation
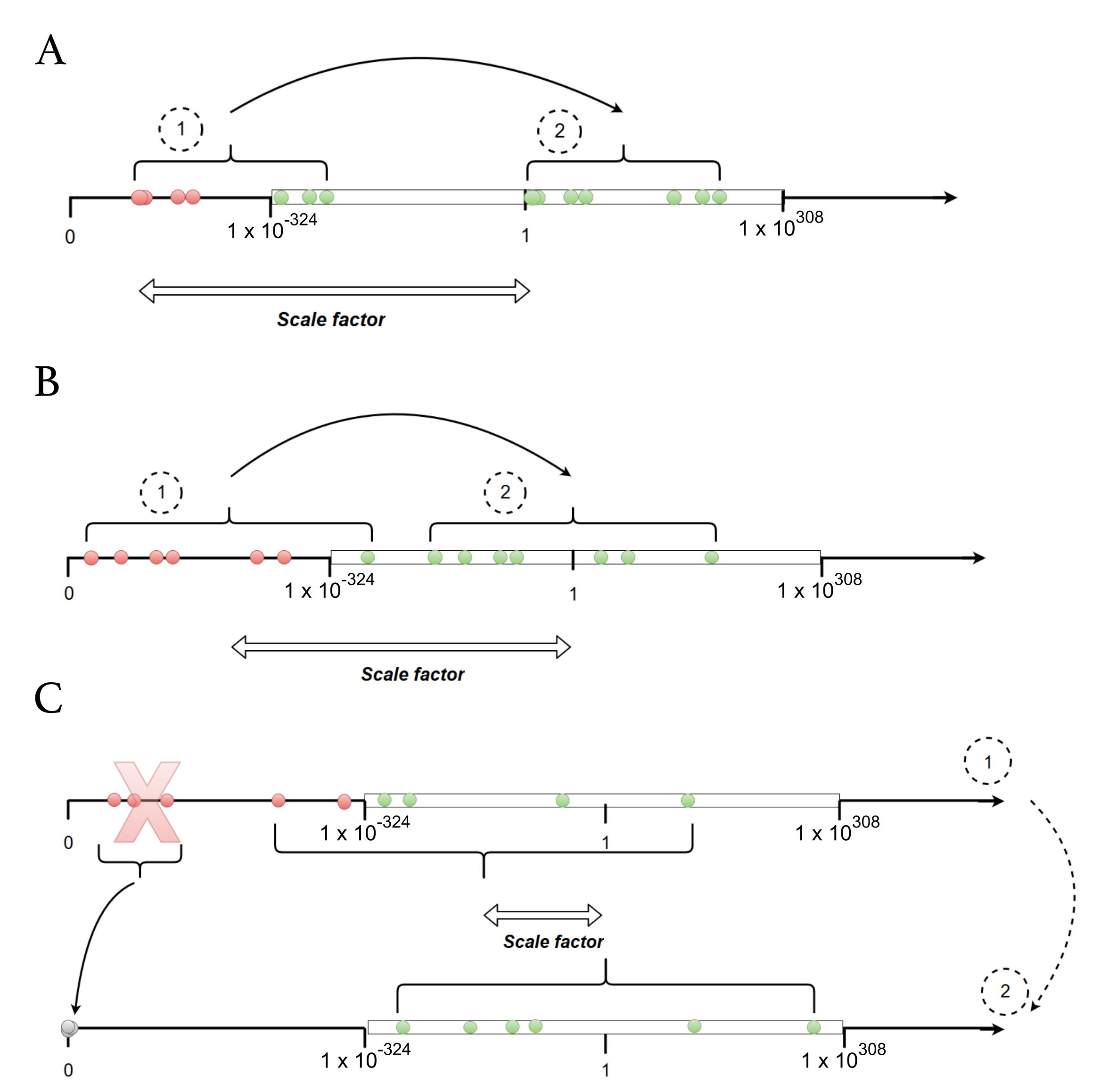
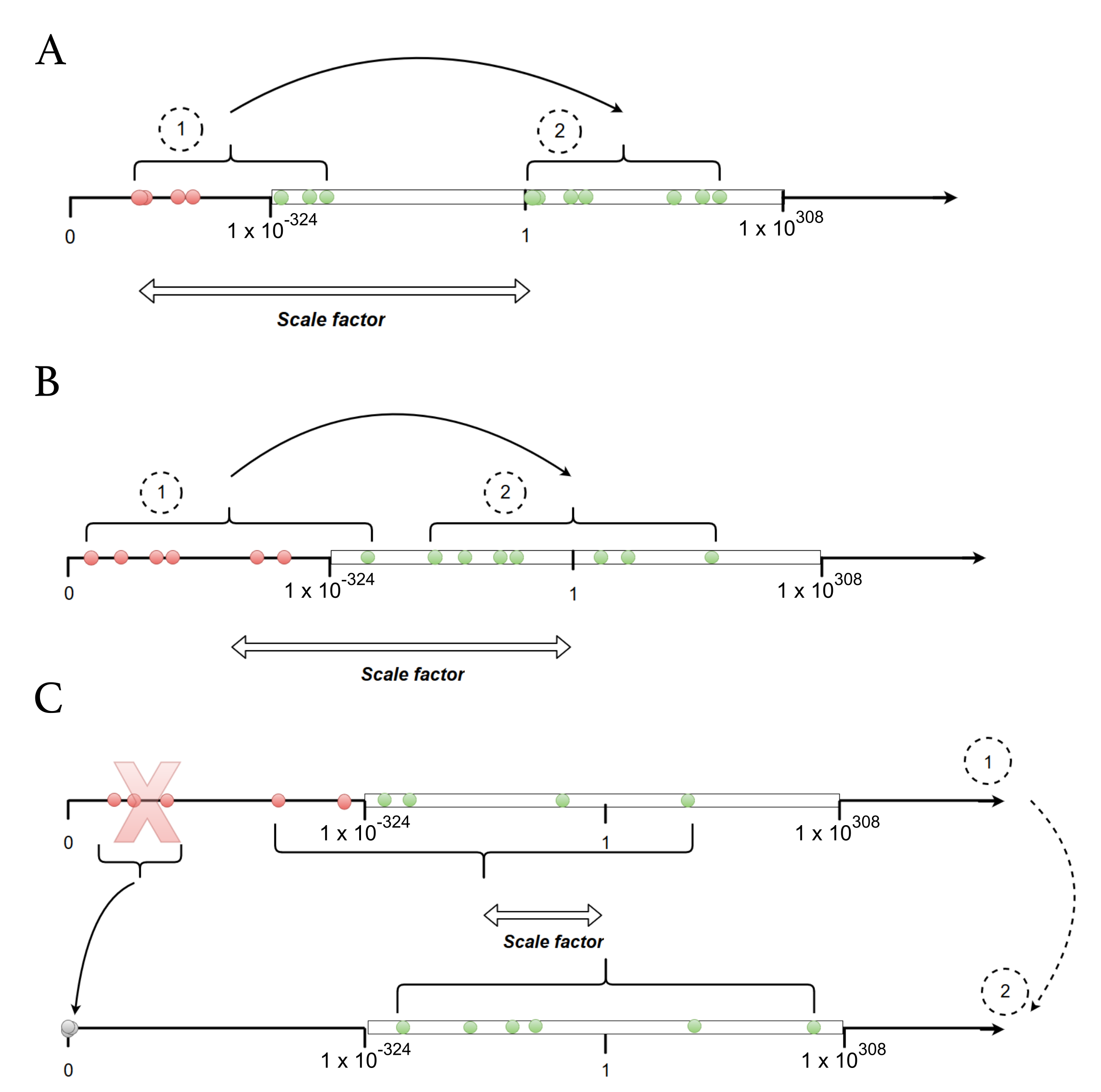
Appendix B.2. Choice of a Scale Factor
Appendix B.3. Performance Improvements
Appendix C. Details on Likelihood Comparisons
Appendix D. Additional Details on Influenza Data Analysis
Appendix E. Tables

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Distribution |
|---|---|
| Unif(1, 3) | |
| Unif(0, 1) | |
| Unif(0, 0.5) | |
| Unif(0.05, 0.5) | |
| Unif(0, 1) |
| Parameter | Value |
|---|---|
| 0.4 | |
| 0.3 | |
| 0.27 | |
| 0.17 | |
| 0.03 | |
| 0.03 | |
| 0.03 | |
| 0.03 | |
| Initial root state | 1 |
| Parameter | Prior Distribution |
|---|---|
| R | LogNormal(0, 1.0) |
| LogNormal(4.5, 0.15) | |
| LogNormal(0, 2.0) | |
| LogNormal(0, 0.5) | |
| s | Exp(0.001) truncated on |
| r | Beta(10.0, 1.5) |
| T | LogNormal(2.0, 1.0) |
| 175 Samples | 500 Samples | |||||
|---|---|---|---|---|---|---|
| m | hpd_low | hpd_high | m | hpd_low | hpd_high | |
| t | 3.342 | 3.048 | 3.643 | 6.645 | 6.313 | 7.031 |
| 90.464 | 75.836 | 105.207 | 101.582 | 87.197 | 116.768 | |
| 0.354 | 0.199 | 0.556 | 0.505 | 0.263 | 0.815 | |
| 0.971 | 0.927 | 1.01 | 1.001 | 0.98 | 1.02 | |
| 1.048 | 1.026 | 1.071 | 1.005 | 0.992 | 1.017 | |
| 0.991 | 0.969 | 1.013 | 1.01 | 0.998 | 1.022 | |
| 0.558 | 0.335 | 0.783 | 0.774 | 0.679 | 0.861 | |
| 1.08 | 1.037 | 1.123 | 1.027 | 1.002 | 1.051 | |
| 0.475 | 0.196 | 0.869 | 0.304 | 0.147 | 0.524 | |
| 0.871 | 0.245 | 1.923 | 1.064 | 0.3 | 2.422 | |
| 0.894 | 0.253 | 1.965 | 0.838 | 0.264 | 1.744 | |
| 2.183 | 0.877 | 4.174 | 1.568 | 0.635 | 2.973 | |
| 0.561 | 0.205 | 1.08 | 0.694 | 0.332 | 1.248 | |
| 1.001 | 0.273 | 2.261 | 0.887 | 0.275 | 1.93 | |
| 1.005 | 0.279 | 2.173 | 1.13 | 0.368 | 2.468 | |
| 0.292 | 0 | 0.777 | 0.296 | 0 | 0.779 | |
| 0.3 | 0 | 0.784 | 0.292 | 0 | 0.777 | |
| 0.283 | 0 | 0.769 | 0.289 | 0 | 0.767 | |
| 0.024 | 0 | 0.535 | 0.023 | 0 | 0.501 | |
| 0.849 | 0.167 | 1 | 0.863 | 0.176 | 1 | |
| 0.041 | 0 | 0.731 | 0.044 | 0 | 0.67 | |
Appendix F. Additional Figures
References
- Felsenstein, J. Estimating effective population size from samples of sequences: Inefficiency of pairwise and segregating sites as compared to phylogenetic estimates. Genet. Res. 1992, 59, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Hey, J.; Machado, C.A. The study of structured populations? New hope for a difficult and divided science. Nat. Rev. Genet. 2003, 4, 535–543. [Google Scholar] [CrossRef]
- Stadler, T.; Bonhoeffer, S. Uncovering epidemiological dynamics in heterogeneous host populations using phylogenetic methods. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2013, 368, 20120198. [Google Scholar] [CrossRef] [PubMed]
- Grenfell, B.T.; Pybus, O.G.; Gog, J.R.; Wood, J.L.; Daly, J.M.; Mumford, J.A.; Holmes, E.C. Unifying the epidemiological and evolutionary dynamics of pathogens. Science 2004, 303, 327–332. [Google Scholar] [CrossRef]
- Kühnert, D.; Wu, C.H.; Drummond, A.J. Phylogenetic and epidemic modeling of rapidly evolving infectious diseases. Infect. Genet. Evol. 2011, 11, 1825–1841. [Google Scholar] [CrossRef] [PubMed]
- Dudas, G.; Carvalho, L.M.; Bedford, T.; Tatem, A.J.; Baele, G.; Faria, N.R.; Park, D.J.; Ladner, J.T.; Arias, A.; Asogun, D.; et al. Virus genomes reveal factors that spread and sustained the Ebola epidemic. Nature 2017, 544, 309. [Google Scholar] [CrossRef] [PubMed]
- Faria, N.R.; Kraemer, M.U.; Hill, S.; De Jesus, J.G.; Aguiar, R.; Iani, F.C.; Xavier, J.; Quick, J.; Du Plessis, L.; Dellicour, S.; et al. Genomic and epidemiological monitoring of yellow fever virus transmission potential. Science 2018, 361, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef]
- Hodges, S.A. Floral nectar spurs and diversification. Int. J. Plant Sci. 1997, 158, S81–S88. [Google Scholar] [CrossRef]
- Goldberg, E.E.; Kohn, J.R.; Lande, R.; Robertson, K.A.; Smith, S.A.; Igić, B. Species selection maintains self-incompatibility. Science 2010, 330, 493–495. [Google Scholar] [CrossRef] [PubMed]
- Mayrose, I.; Zhan, S.H.; Rothfels, C.J.; Magnuson-Ford, K.; Barker, M.S.; Rieseberg, L.H.; Otto, S.P. Recently formed polyploid plants diversify at lower rates. Science 2011, 333, 1257. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, E.E.; Lancaster, L.T.; Ree, R.H. Phylogenetic inference of reciprocal effects between geographic range evolution and diversification. Syst. Biol. 2011, 60, 451–465. [Google Scholar] [CrossRef]
- Volz, E.M.; Frost, S.D. Sampling through time and phylodynamic inference with coalescent and birth–death models. J. R. Soc. Interface 2014, 11, 20140945. [Google Scholar] [CrossRef] [PubMed]
- Boskova, V.; Bonhoeffer, S.; Stadler, T. Inference of epidemiological dynamics based on simulated phylogenies using birth-death and coalescent models. PLoS Comput. Biol. 2014, 10, e1003913. [Google Scholar] [CrossRef]
- Maddison, W.P.; Midford, P.E.; Otto, S.P. Estimating a binary character’s effect on speciation and extinction. Syst. Biol. 2007, 56, 701–710. [Google Scholar] [CrossRef]
- Kühnert, D.; Stadler, T.; Vaughan, T.G.; Drummond, A.J. Phylodynamics with Migration: A Computational Framework to Quantify Population Structure from Genomic Data. Mol. Biol. Evol. 2016, 33, 2102–2116. [Google Scholar] [CrossRef]
- Stadler, T.; Kühnert, D.; Bonhoeffer, S.; Drummond, A.J. Birth-death skyline plot reveals temporal changes of epidemic spread in HIV and hepatitis C virus (HCV). Proc. Natl. Acad. Sci. USA 2013, 110, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, T.G.; Kühnert, D.; Popinga, A.; Welch, D.; Drummond, A.J. Efficient Bayesian inference under the structured coalescent. Bioinformatics 2014, 30, 2272–2279. [Google Scholar] [CrossRef]
- Gavryushkina, A.; Welch, D.; Stadler, T.; Drummond, A.J. Bayesian inference of sampled ancestor trees for epidemiology and fossil calibration. PLoS Comput. Biol. 2014, 10, e1003919. [Google Scholar] [CrossRef]
- Louca, S.; Pennell, M.W. Extant timetrees are consistent with a myriad of diversification histories. Nature 2020, 580, 502–505. [Google Scholar] [CrossRef] [PubMed]
- Louca, S.; McLaughlin, A.; MacPherson, A.; Joy, J.B.; Pennell, M.W. Fundamental identifiability limits in molecular epidemiology. Mol. Biol. Evol. 2021, 38, 4010–4024. [Google Scholar] [CrossRef]
- MacPherson, A.; Louca, S.; McLaughlin, A.; Joy, J.B.; Pennell, M.W. Unifying Phylogenetic Birth–Death Models in Epidemiology and Macroevolution. Syst. Biol. 2022, 71, 172–189. [Google Scholar] [CrossRef]
- Maddison, W.P. Confounding asymmetries in evolutionary diversification and character change. Evolution 2006, 60, 1743–1746. [Google Scholar] [CrossRef]
- FitzJohn, R.G. Diversitree: Comparative phylogenetic analyses of diversification in R. Methods Ecol. Evol. 2012, 3, 1084–1092. [Google Scholar] [CrossRef]
- Rabosky, D.L.; Goldberg, E.E. Model inadequacy and mistaken inferences of trait-dependent speciation. Syst. Biol. 2015, 64, 340–355. [Google Scholar] [CrossRef] [PubMed]
- Dormand, J.R.; Prince, P.J. A family of embedded Runge-Kutta formulae. J. Comput. Appl. Math. 1980, 6, 19–26. [Google Scholar] [CrossRef]
- Math, C. The Apache Commons Mathematics Library. 2016. Available online: https://commons.apache.org/proper/commons-math/ (accessed on 26 July 2022).
- Vaughan, T.G.; Drummond, A.J. A stochastic simulator of birth–death master equations with application to phylodynamics. Mol. Biol. Evol. 2013, 30, 1480–1493. [Google Scholar] [CrossRef]
- Lanave, C.; Preparata, G.; Sacone, C.; Serio, G. A new method for calculating evolutionary substitution rates. J. Mol. Evol. 1984, 20, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchêne, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kühnert, D.; De Maio, N.; et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2019, 15, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.Y. ggtree: An r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scire, J.; Barido-Sottani, J.; Kühnert, D.; Vaughan, T.G.; Stadler, T. Robust Phylodynamic Analysis of Genetic Sequencing Data from Structured Populations. Viruses 2022, 14, 1648. https://doi.org/10.3390/v14081648
Scire J, Barido-Sottani J, Kühnert D, Vaughan TG, Stadler T. Robust Phylodynamic Analysis of Genetic Sequencing Data from Structured Populations. Viruses. 2022; 14(8):1648. https://doi.org/10.3390/v14081648
Chicago/Turabian StyleScire, Jérémie, Joëlle Barido-Sottani, Denise Kühnert, Timothy G. Vaughan, and Tanja Stadler. 2022. "Robust Phylodynamic Analysis of Genetic Sequencing Data from Structured Populations" Viruses 14, no. 8: 1648. https://doi.org/10.3390/v14081648
APA StyleScire, J., Barido-Sottani, J., Kühnert, D., Vaughan, T. G., & Stadler, T. (2022). Robust Phylodynamic Analysis of Genetic Sequencing Data from Structured Populations. Viruses, 14(8), 1648. https://doi.org/10.3390/v14081648