
Genomic Diversity of Torque Teno Virus in Blood Samples from Febrile Paediatric Outpatients in Tanzania: A Descriptive Cohort Study


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Origin of mNGS Raw Data Used in This Study
2.2. Bioinformatic Analysis
2.2.1. Database
2.2.2. Mapping
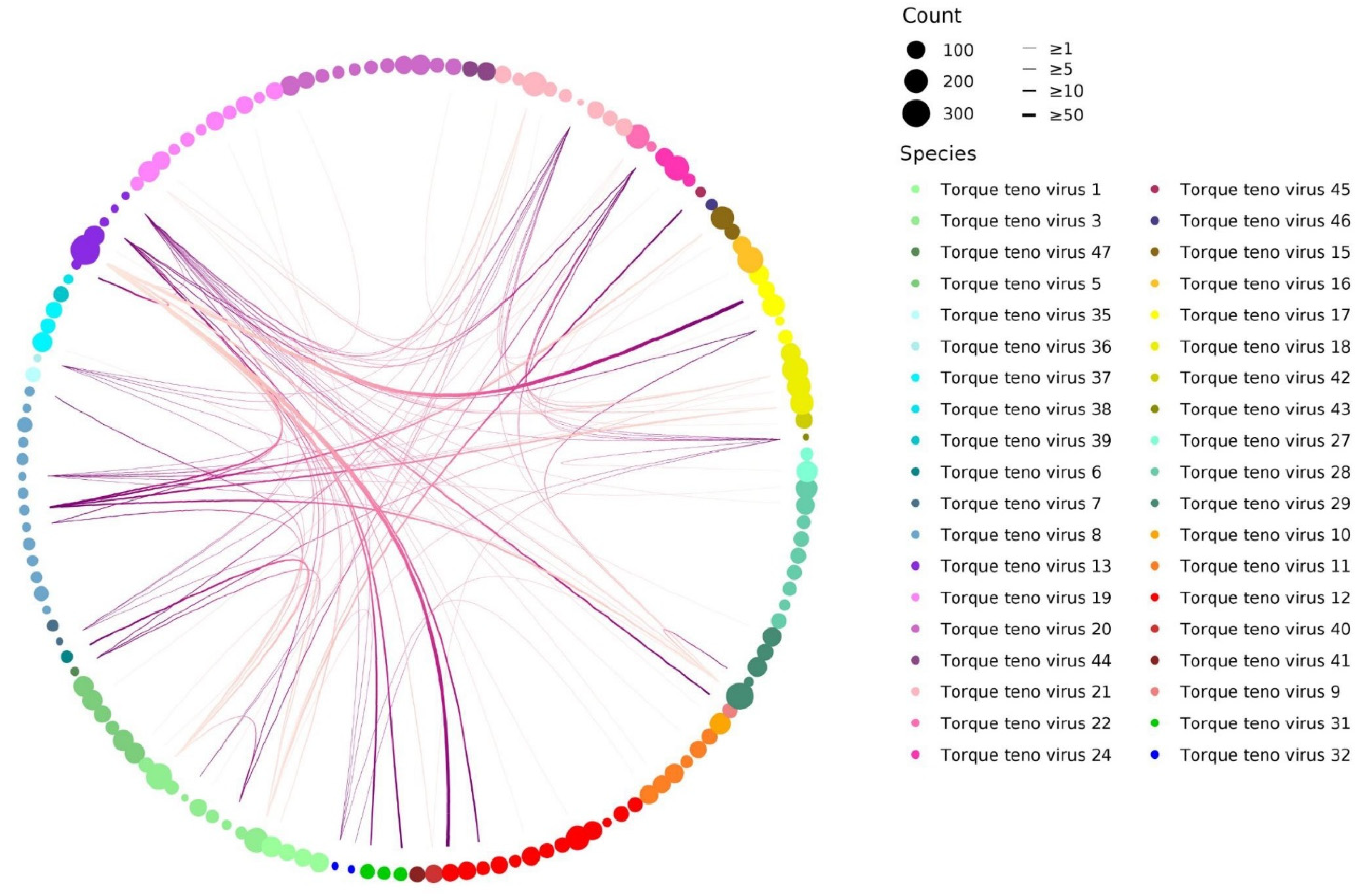
2.3. Circos Plot
2.4. Statistical Analyses
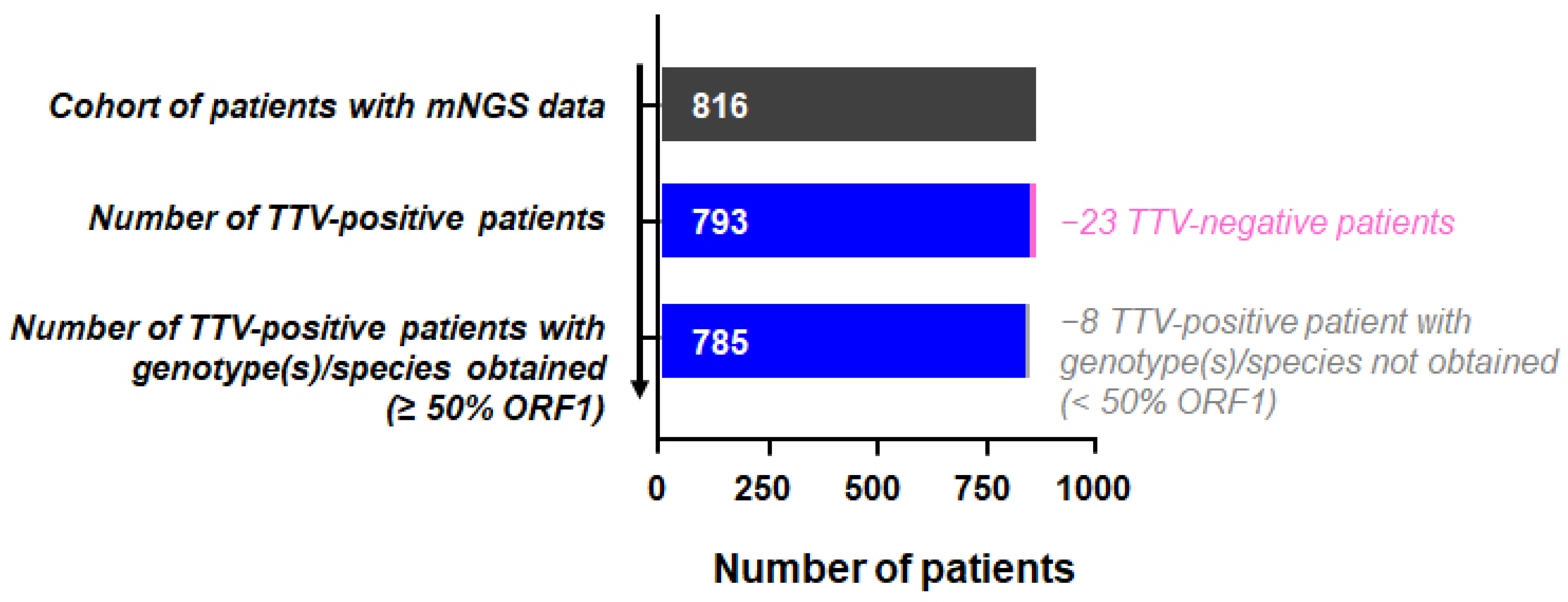
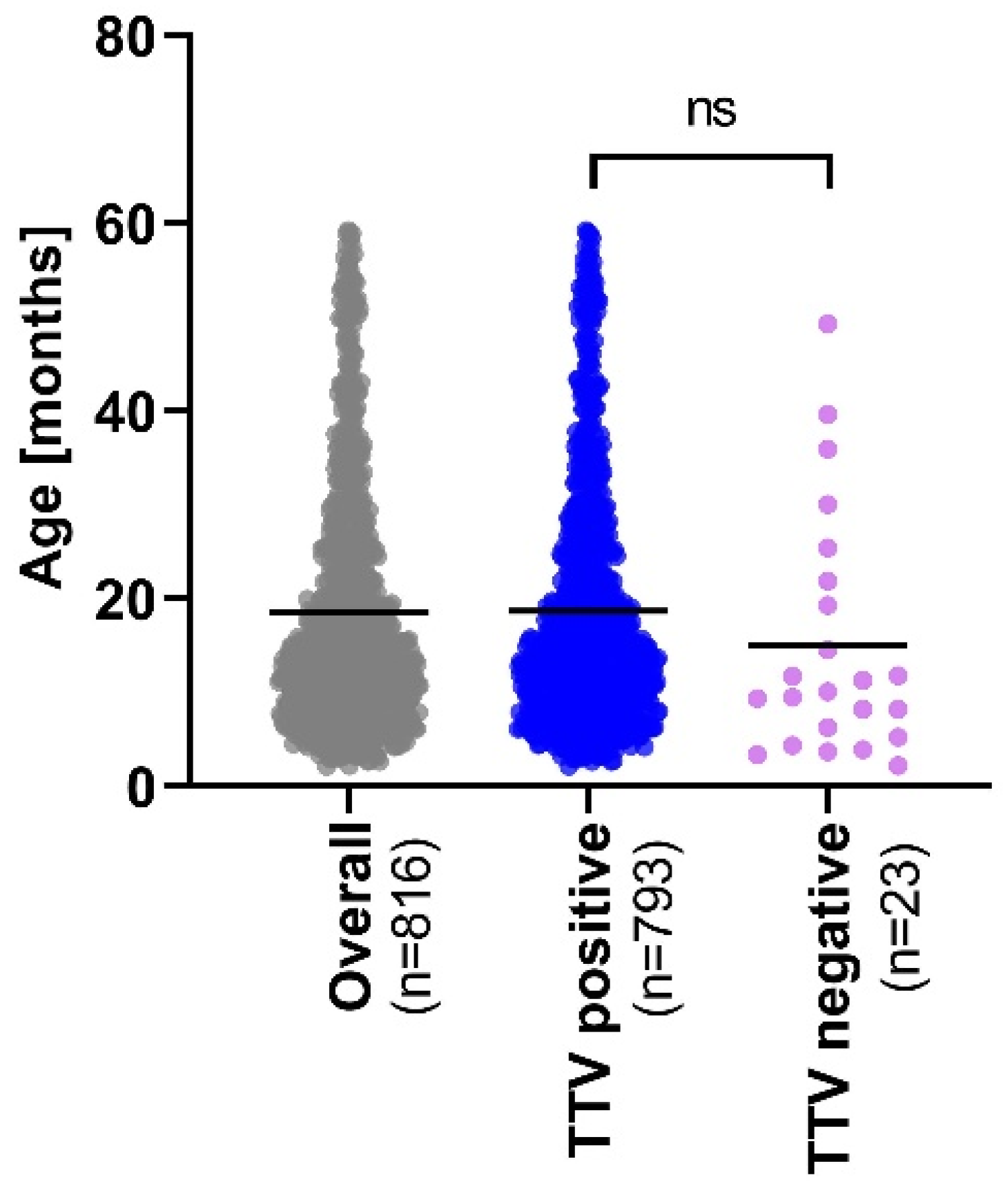
3. Results
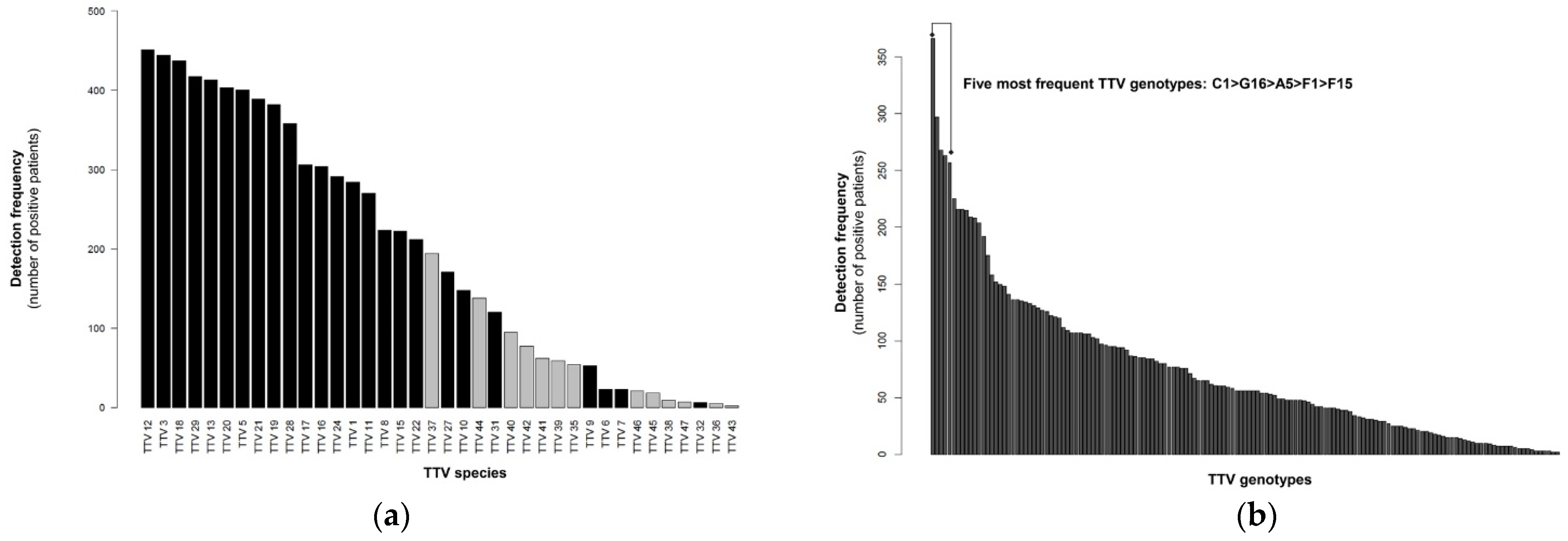
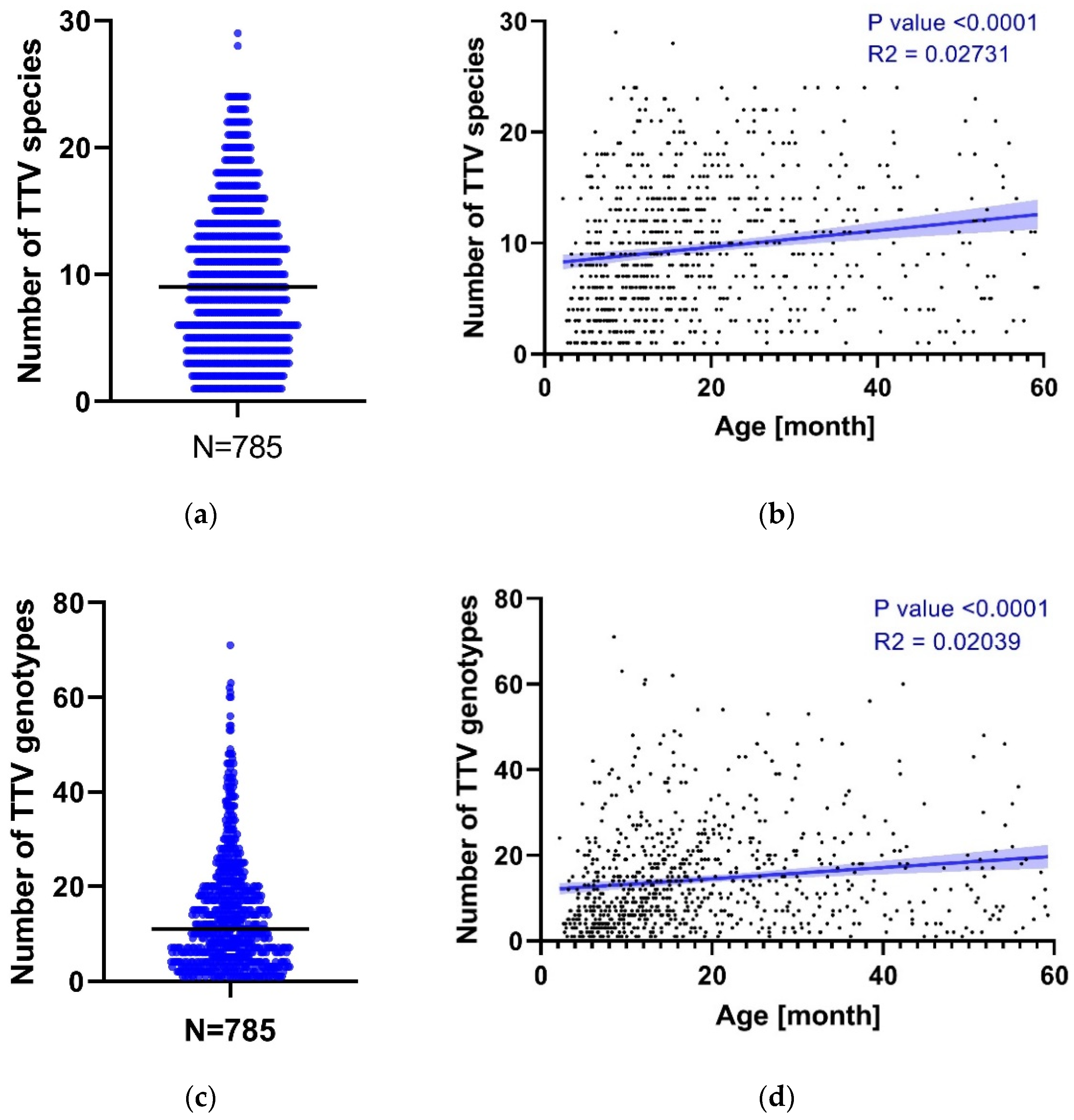
3.1. TTV Genomic Diversity
3.2. TTV Species and Genotype Co-Detections
3.3. TTV Genotypes Relationship Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Varsani, A.; Opriessnig, T.; Celer, V.; Maggi, F.; Okamoto, H.; Blomström, A.L.; Cadar, D.; Harrach, B.; Biagini, P.; Kraberger, S. Taxonomic update for mammalian anelloviruses (family Anelloviridae). Arch. Virol. 2021, 166, 2943–2953. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, T.; Okamoto, H.; Konishi, K.; Yoshizawa, H.; Miyakawa, Y.; Mayumi, M. A novel DNA virus (TTV) associated with elevated transaminase levels in posttransfusion hepatitis of unknown etiology. Biochem. Biophys. Res. Commun. 1997, 241, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Zanella, M.C.; Cordey, S.; Kaiser, L. Beyond Cytomegalovirus and Epstein-Barr Virus: A Review of Viruses Composing the Blood Virome of Solid Organ Transplant and Hematopoietic Stem Cell Transplant Recipients. Clin. Microbiol. Rev. 2020, 33, e00027-20. [Google Scholar] [CrossRef] [PubMed]
- Kaczorowska, J.; van der Hoek, L. Human anelloviruses: Diverse, omnipresent and commensal members of the virome. FEMS Microbial. Rev. 2020, 44, 305–313. [Google Scholar] [CrossRef] [Green Version]
- Lolomadze, E.A.; Rebrikov, D.V. Constant companion: Clinical and developmental aspects of torque teno virus infections. Arch. Virol. 2020, 165, 2749–2757. [Google Scholar] [CrossRef]
- Spandole, S.; Cimponeriu, D.; Berca, L.M.; Mihăescu, G. Human anelloviruses: An update of molecular, epidemiological and clinical aspects. Arch. Virol. 2015, 160, 893–908. [Google Scholar] [CrossRef]
- Maggi, F.; Focosi, D.; Albani, M.; Lanini, L.; Vatteroni, M.L.; Petrini, M.; Ceccherini-Nelli, L.; Pistello, M.; Bendinelli, M. Role of hematopoietic cells in the maintenance of chronic human torquetenovirus plasma viremia. J. Virol. 2010, 84, 6891–6893. [Google Scholar] [CrossRef] [Green Version]
- Mariscal, L.F.; López-Alcorocho, J.M.; Rodríguez-Iñigo, E.; Ortiz-Movilla, N.; de Lucas, S.; Bartolomé, J.; Carreño, V. TT virus replicates in stimulated but not in nonstimulated peripheral blood mononuclear cells. Virology 2002, 301, 121–129. [Google Scholar] [CrossRef] [Green Version]
- Maggi, F.; Pistello, M.; Vatteroni, M.; Presciuttini, S.; Marchi, S.; Isola, P.; Fornai, C.; Fagnani, S.; Andreoli, E.; Antonelli, G.; et al. Dynamics of persistent TT virus infection, as determined in patients treated with alpha interferon for concomitant hepatitis C virus infection. J. Virol. 2001, 75, 11999–12004. [Google Scholar] [CrossRef] [Green Version]
- Lim, E.S.; Zhou, Y.; Zhao, G.; Bauer, I.K.; Droit, L.; Ndao, I.M.; Warner, B.B.; Tarr, P.I.; Wang, D.; Holtz, L.R. Early life dynamics of the human gut virome and bacterial microbiome in infants. Nat. Med. 2015, 21, 1228–1234. [Google Scholar] [CrossRef]
- Ohto, H.; Ujiie, N.; Takeuchi, C.; Sato, A.; Hayashi, A.; Ishiko, H.; Nishizawa, T.; Okamoto, H. TT virus infection during childhood. Transfusion 2002, 42, 892–898. [Google Scholar] [CrossRef] [PubMed]
- Tyschik, E.A.; Rasskazova, A.S.; Degtyareva, A.V.; Rebrikov, D.V.; Sukhikh, G.T. Torque teno virus dynamics during the first year of life. Virol. J. 2018, 15, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagaglio, S.; Sitia, G.; Prati, D.; Cella, D.; Hasson, H.; Novati, R.; Lazzarin, A.; Morsica, G. Mother-to-child transmission of TT virus: Sequence analysis of non-coding region of TT virus in infected mother-infant pairs. Arch. Virol. 2002, 147, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, H.; Inui, A.; Sogo, T.; Kuroda, K.; Tanaka, T.; Fujisawa, T. TTV infection in children born to mothers infected with TTV but not with HBV, HCV, or HIV. J. Med. Virol. 2004, 74, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Yokozaki, S.; Fukuda, Y.; Nakano, I.; Katano, Y. TT virus: A mother-to-child transmitted rather than bloodborne virus. Blood 1999, 93, 3569–3570. [Google Scholar] [CrossRef]
- Arze, C.A.; Springer, S.; Dudas, G.; Patel, S.; Bhattacharyya, A.; Swaminathan, H.; Brugnara, C.; Delagrave, S.; Ong, T.; Kahvejian, A.; et al. Global genome analysis reveals a vast and dynamic anellovirus landscape within the human virome. Cell. Host. Microb. 2021, 29, 1305–1315.e6. [Google Scholar] [CrossRef]
- Bal, A.; Sarkozy, C.; Josset, L.; Cheynet, V.; Oriol, G.; Becker, J.; Vilchez, G.; Sesques, P.; Mallet, F.; Pachot, A.; et al. Metagenomic Next-Generation Sequencing Reveals Individual Composition and Dynamics of Anelloviruses during Autologous Stem Cell Transplant Recipient Management. Viruses 2018, 10, 633. [Google Scholar] [CrossRef] [Green Version]
- Kulifaj, D.; Tilloy, V.; Scaon, E.; Guerin, E.; Essig, M.; Pichon, N.; Hantz, S.; De Bernardi, A.; Joannes, M.; Barranger, C.; et al. Viral metagenomics analysis of kidney donors and recipients: Torque teno virus genotyping and prevalence. J. Med. Virol. 2020, 92, 3301–3311. [Google Scholar] [CrossRef]
- Kaczorowska, J.; Deijs, M.; Klein, M.; Bakker, M.; Jebbink, M.F.; Sparreboom, M.; Kinsella, C.M.; Timmerman, A.L.; van der Hoek, L. Diversity and Long-Term Dynamics of Human Blood Anelloviruses. J. Virol. 2022, 96, e0010922. [Google Scholar] [CrossRef]
- Fahsbender, E.; Burns, J.M.; Kim, S.; Kraberger, S.; Frankfurter, G.; Eilers, A.A.; Shero, M.R.; Beltran, R.; Kirkham, A.; McCorkell, R.; et al. Diverse and highly recombinant anelloviruses associated with Weddell seals in Antarctica. Virus Evol. 2017, 3, vex017. [Google Scholar] [CrossRef] [Green Version]
- Manni, F.; Rotola, A.; Caselli, E.; Bertorelle, G.; Di Luca, D. Detecting recombination in TT virus: A phylogenetic approach. J. Mol. Evol. 2002, 55, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Worobey, M. Extensive homologous recombination among widely divergent TT viruses. J. Virol. 2000, 74, 7666–7670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordey, S.; Laubscher, F.; Hartley, M.A.; Junier, T.; Keitel, K.; Docquier, M.; Guex, N.; Iseli, C.; Vieille, G.; Le Mercier, P.; et al. Blood virosphere in febrile Tanzanian children. Emerg. Microbes Infect. 2021, 10, 982–993. [Google Scholar] [CrossRef] [PubMed]
- Keitel, K.; Kagoro, F.; Samaka, J.; Masimba, J.; Said, Z.; Temba, H.; Mlaganile, T.; Sangu, W.; Rambaud-Althaus, C.; Gervaix, A.; et al. A novel electronic algorithm using host biomarker point-of-care tests for the management of febrile illnesses in Tanzanian children (e-POCT): A randomized, controlled non-inferiority trial. PLoS Med. 2017, 14, e1002411. [Google Scholar] [CrossRef]
- Van Doorslaer, K. Evolution of the papillomaviridae. Virology 2013, 445, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laubscher, F.; Hartley, M.-A.; Kaiser, L.; Cordey, S. Genomic Diversity of Torque Teno Virus in Blood Samples from Febrile Paediatric Outpatients in Tanzania: A Descriptive Cohort Study. Viruses 2022, 14, 1612. https://doi.org/10.3390/v14081612
Laubscher F, Hartley M-A, Kaiser L, Cordey S. Genomic Diversity of Torque Teno Virus in Blood Samples from Febrile Paediatric Outpatients in Tanzania: A Descriptive Cohort Study. Viruses. 2022; 14(8):1612. https://doi.org/10.3390/v14081612
Chicago/Turabian StyleLaubscher, Florian, Mary-Anne Hartley, Laurent Kaiser, and Samuel Cordey. 2022. "Genomic Diversity of Torque Teno Virus in Blood Samples from Febrile Paediatric Outpatients in Tanzania: A Descriptive Cohort Study" Viruses 14, no. 8: 1612. https://doi.org/10.3390/v14081612
APA StyleLaubscher, F., Hartley, M.-A., Kaiser, L., & Cordey, S. (2022). Genomic Diversity of Torque Teno Virus in Blood Samples from Febrile Paediatric Outpatients in Tanzania: A Descriptive Cohort Study. Viruses, 14(8), 1612. https://doi.org/10.3390/v14081612