
Pseudorabies Virus Tegument Protein UL13 Suppresses RLR-Mediated Antiviral Innate Immunity through Regulating Receptor Transcription


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Antibodies and Reagents
2.3. Lentiviral Infection and Stable Cell Line Generation
2.4. Reverse Transcription and Quantitative Real-Time PCR (qPCR)
2.5. Immunoblotting
2.6. Dual-Luciferase Reporter Assay
2.7. Inhibition of Signaling Pathways
2.8. Cytoplasmic and Nuclear Protein Extraction
2.9. Immunofluorescence
2.10. Statistical Analysis
3. Results
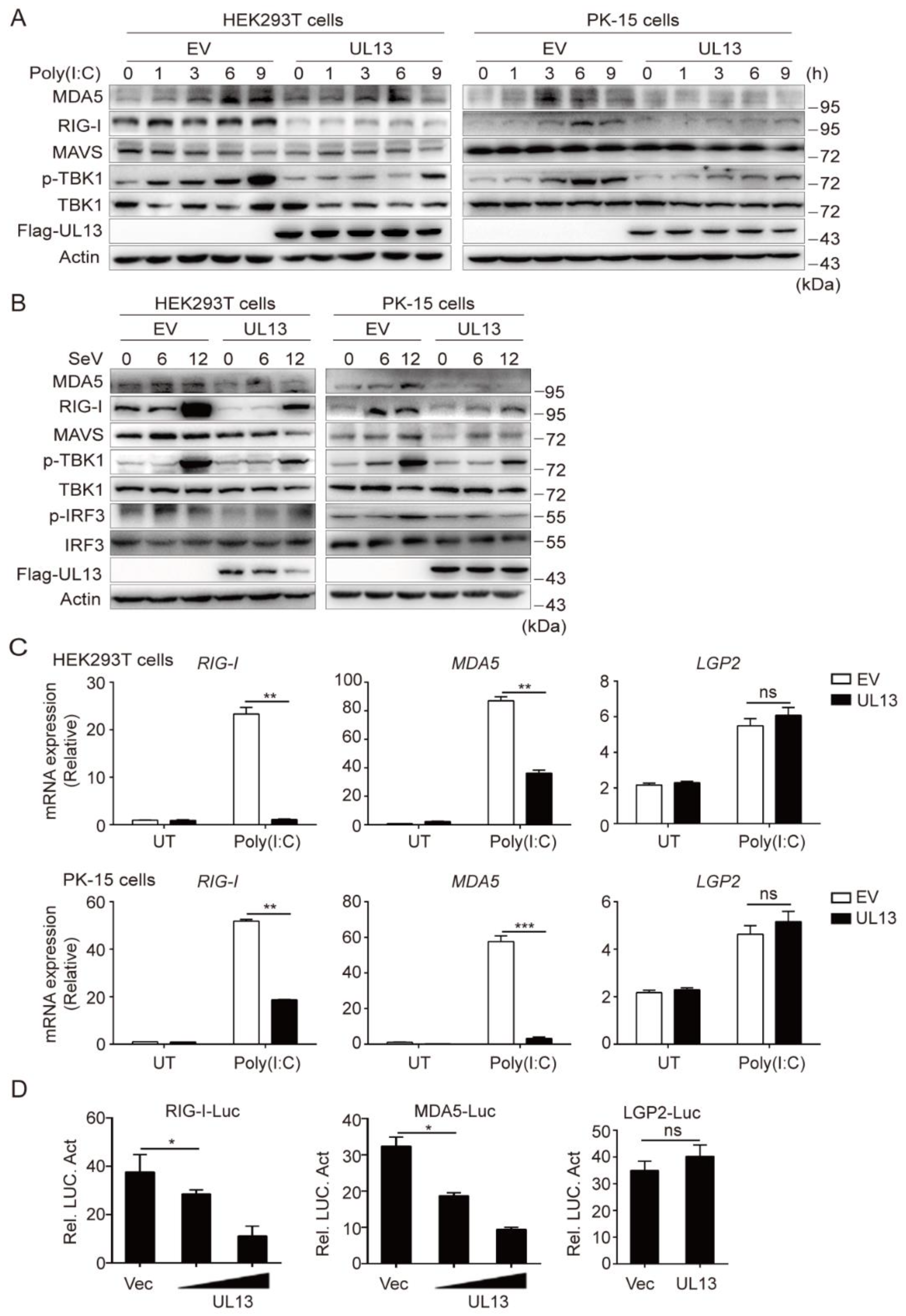
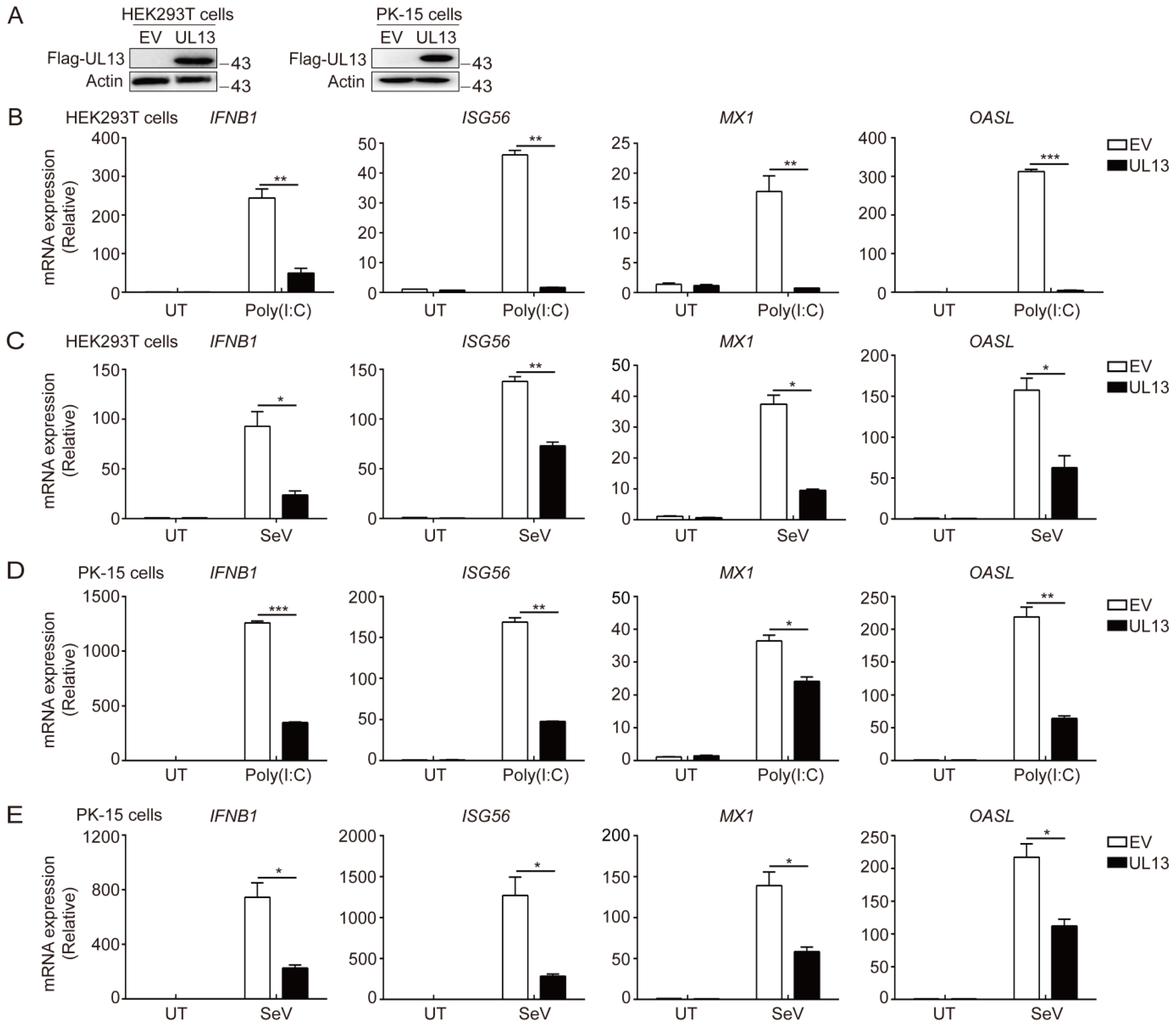
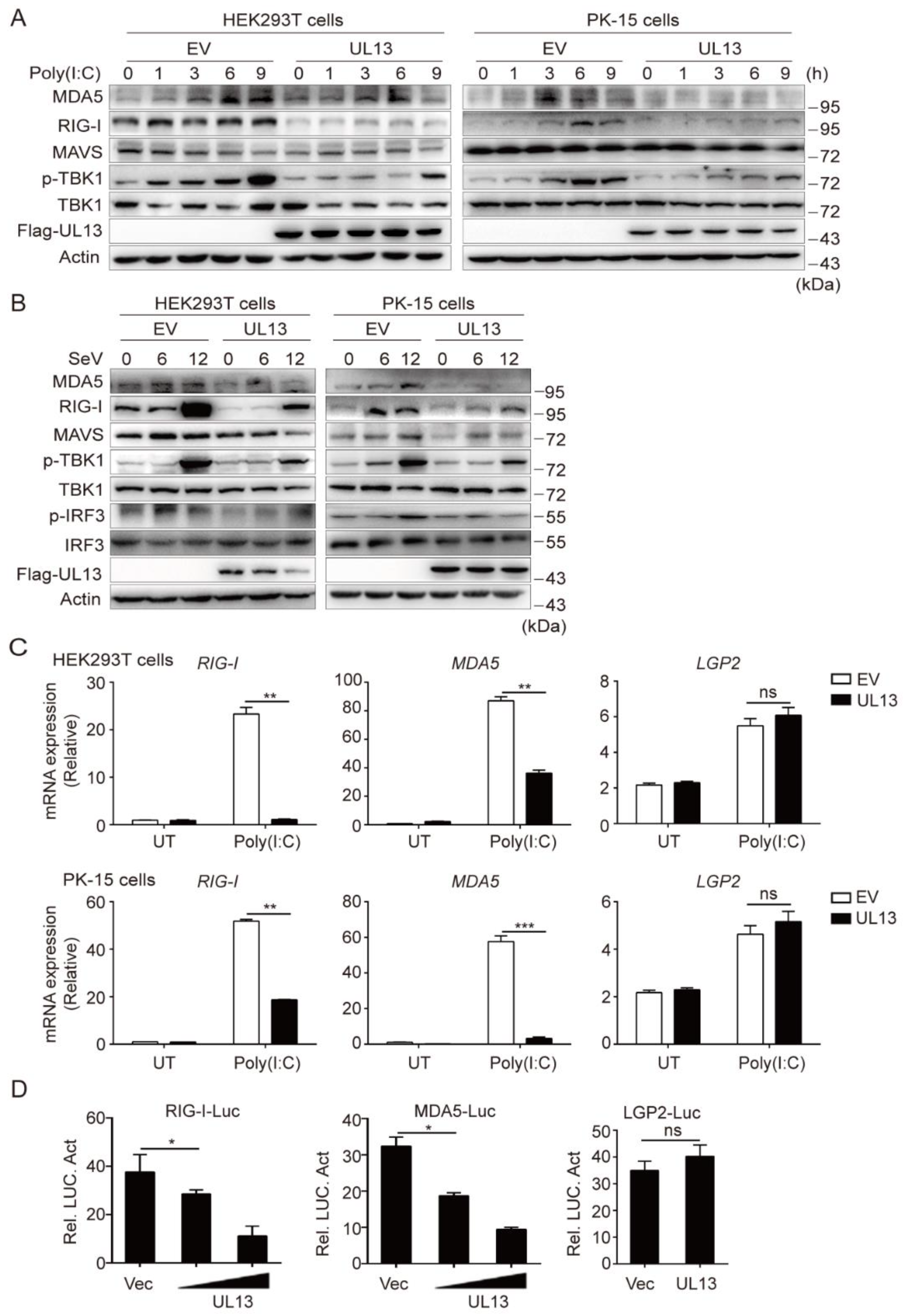
3.1. UL13 Inhibits RLR-mediated Antiviral Immune Responses
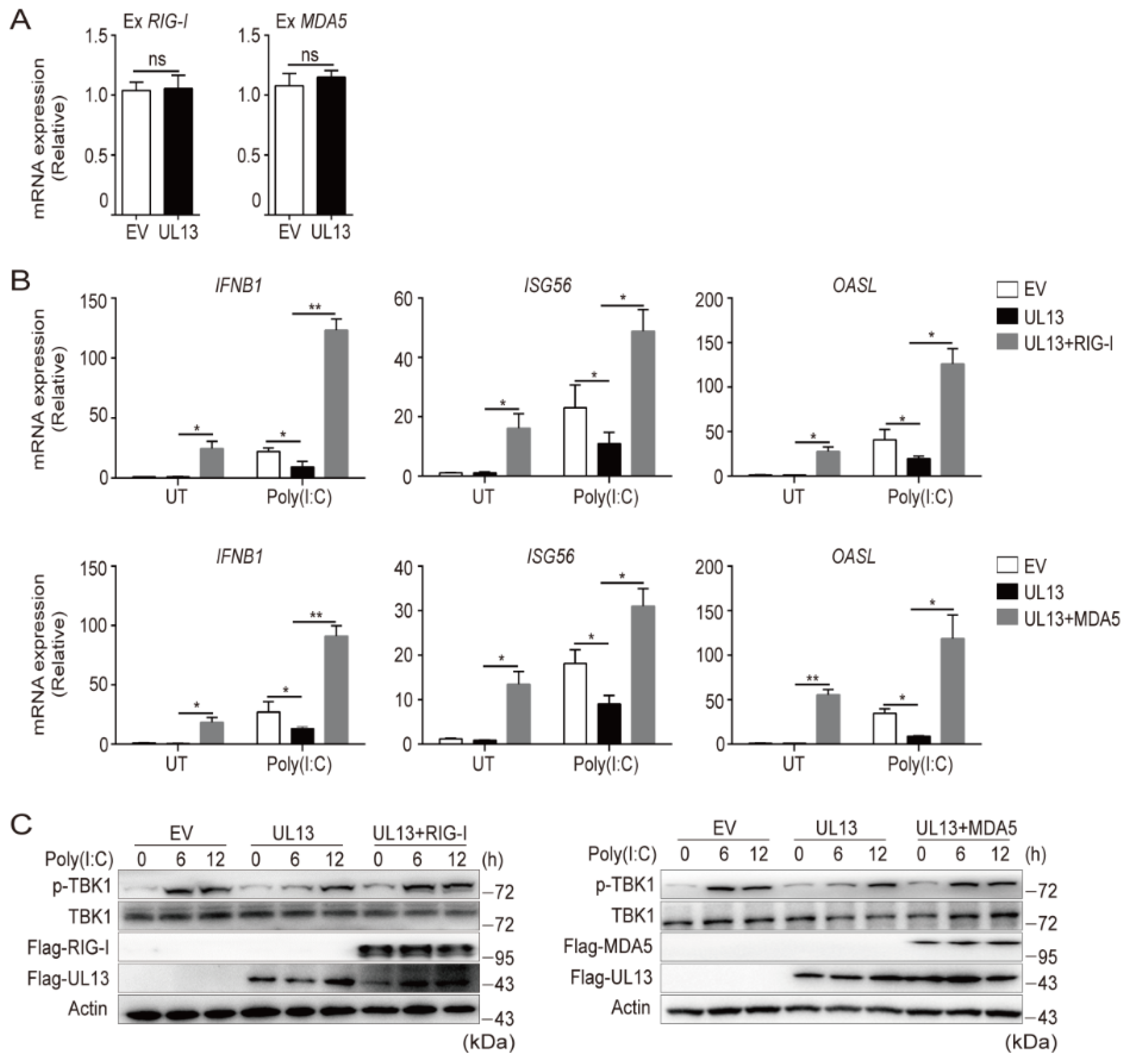
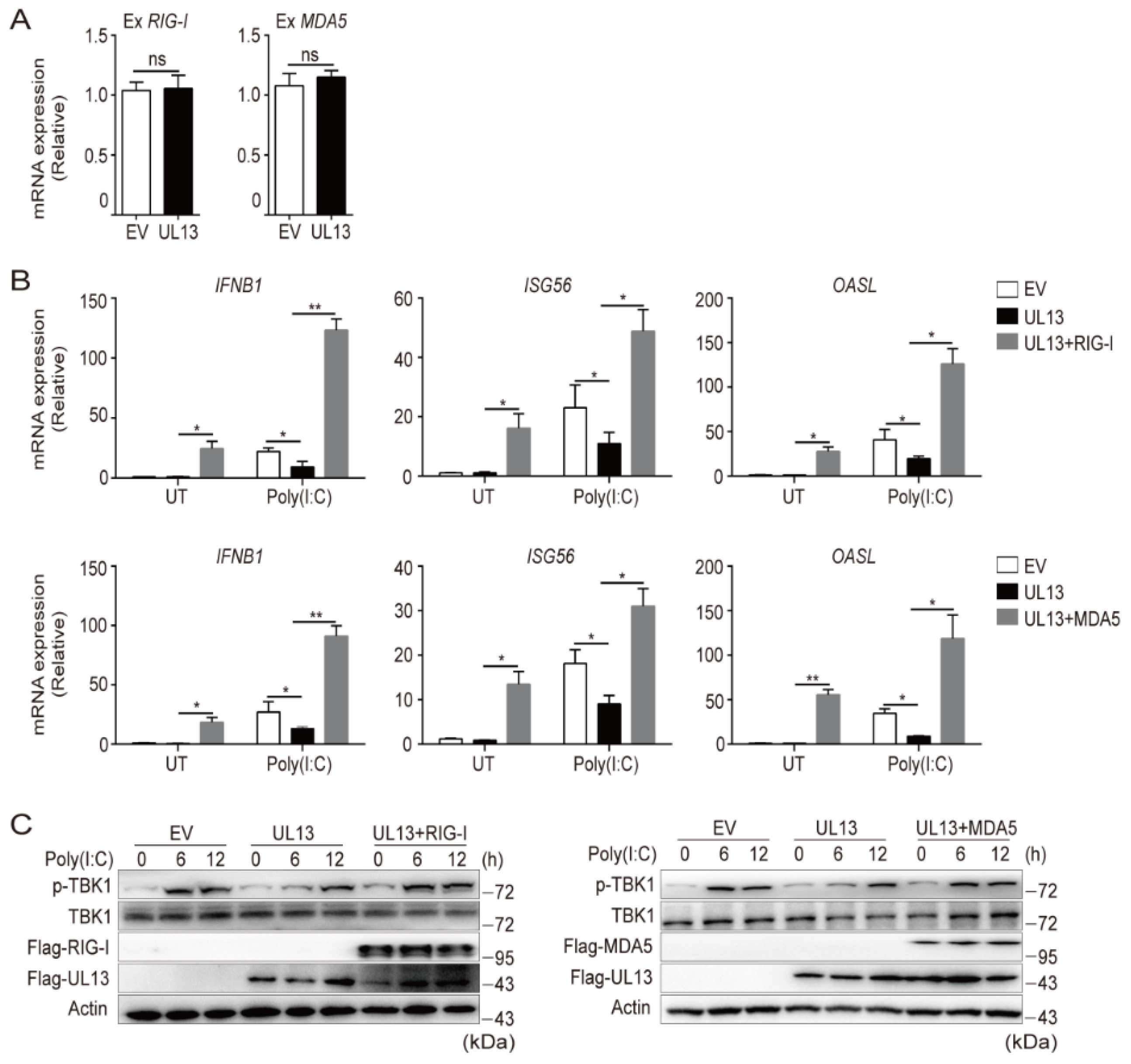
3.2. UL13 Inhibits RLR-Mediated Antiviral Response by Suppressing Transcription of RIG-I and MDA5
3.3. Enforced Expression of RIG-I and MDA5 Counteracts UL13-Suppressed Antiviral Immune Responses
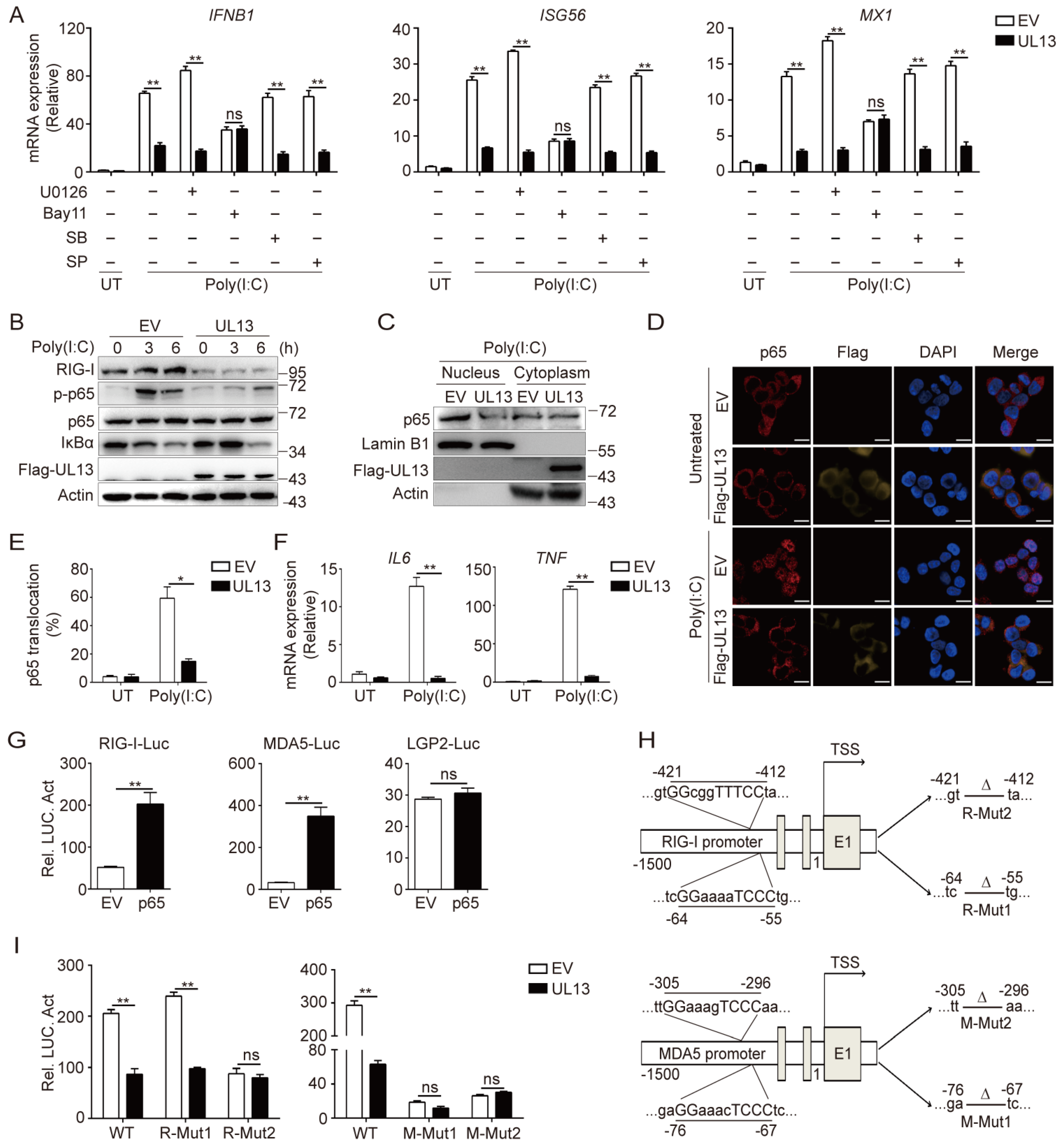
3.4. UL13 Overexpression Restrains NF-κB Activation to Modulate Transcription of RIG-I and MDA5
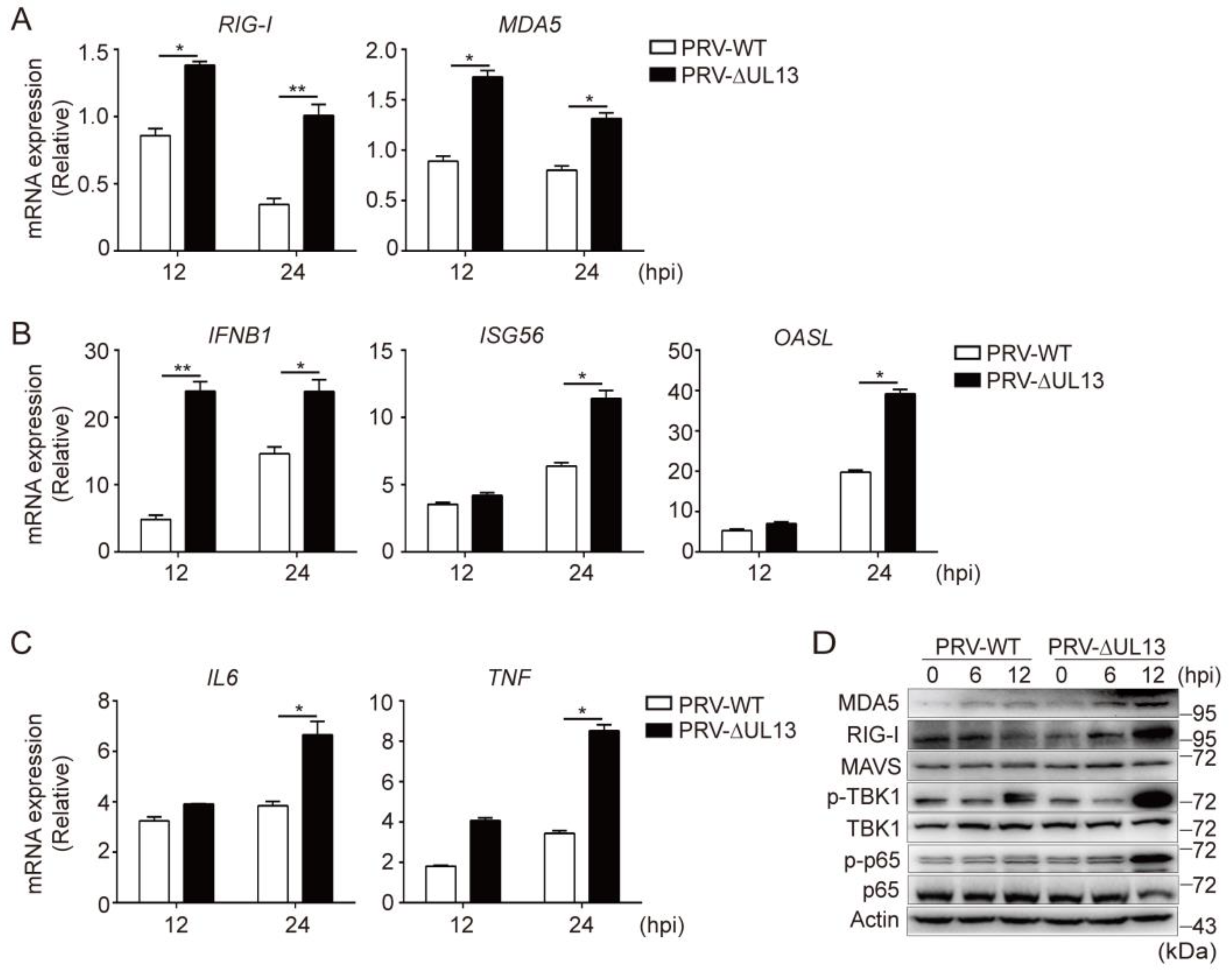
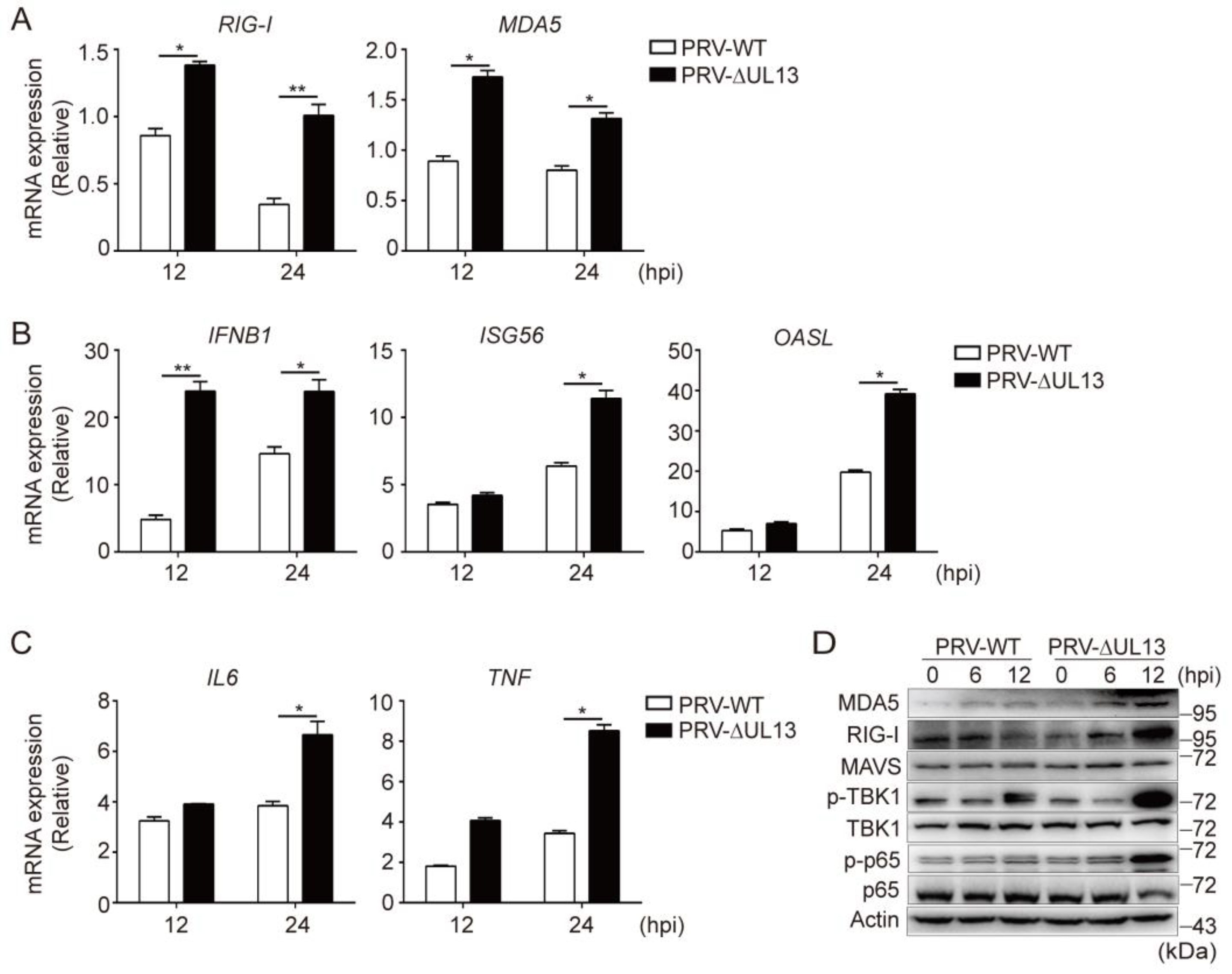
3.5. UL13 Deficiency Potentiates RLR-Mediated Antiviral Responses during PRV Infection
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Laval, K.; Enquist, L.W. The Neuropathic Itch Caused by Pseudorabies Virus. Pathogens 2020, 9, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Liang, R.; Pang, Y.; Shi, L.; Cui, S.; Lin, W. Evidence for interspecies transmission route of pseudorabies virus via virally contaminated fomites. Vet. Microbiol. 2020, 251, 108912. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, C.; Li, X. Novel Chinese pseudorabies virus variants undergo extensive recombination and rapid interspecies transmission. Transbound. Emerg. Dis. 2020, 67, 2274–2276. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wang, X.; Xie, C.; Ding, S.; Yang, H.; Guo, S.; Li, J.; Qin, L.; Ban, F.; Wang, D.; et al. A novel human acute encephalitis caused by pseudorabies virus variant strain. Clin. Infect. Dis. 2020, 73, e3690–e3700. [Google Scholar] [CrossRef] [PubMed]
- Ai, J.-W.; Weng, S.-S.; Cheng, Q.; Cui, P.; Li, Y.-J.; Wu, H.-L.; Zhu, Y.-M.; Xu, B.; Zhang, W.-H. Human Endophthalmitis Caused By Pseudorabies Virus Infection, China, 2017. Emerg. Infect. Dis. 2018, 24, 1087–1090. [Google Scholar] [CrossRef] [Green Version]
- Fan, S.; Yuan, H.; Liu, L.; Li, H.; Wang, S.; Zhao, W.; Wu, Y.; Wang, P.; Hu, Y.; Han, J.; et al. Pseudorabies virus encephalitis in humans: A case series study. J. Neurovirol. 2020, 26, 556–564. [Google Scholar] [CrossRef]
- Zhang, R.; Tang, J. Evasion of I interferon-mediated innate immunity by pseudorabies virus. Front. Microbiol. 2021, 12, 801257. [Google Scholar] [CrossRef]
- Lu, M.; Qiu, S.; Zhang, L.; Sun, Y.; Bao, E.; Lv, Y. Pseudorabies virus glycoprotein gE suppresses interferon-β production via CREB-binding protein degradation. Virus Res. 2020, 291, 198220. [Google Scholar] [CrossRef]
- Xie, J.; Zhang, X.; Chen, L.; Bi, Y.; Idris, A.; Xu, S.; Li, X.; Zhang, Y.; Feng, R. Pseudorabies virus US3 protein inhibits IFN-β production by interacting with IRF3 to block its activation. Front. Microbiol. 2021, 12, 761282. [Google Scholar] [CrossRef]
- Koyanagi, N.; Kawaguchi, Y. Evasion of the Cell-Mediated Immune Response by Alphaherpesviruses. Viruses 2020, 12, 1354. [Google Scholar] [CrossRef]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Stok, J.E.; Vega Quiroz, M.E.; van der Veen, A.G. Self RNA Sensing by RIG-I-like Receptors in Viral Infection and Sterile Inflammation. J. Immunol. 2020, 205, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Bai, X.C.; Chen, Z.J. Structures and Mechanisms in the cGAS-STING Innate Immunity Pathway. Immunity 2020, 53, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Onomoto, K.; Onoguchi, K.; Yoneyama, M. Regulation of RIG-I-like receptor-mediated signaling: Interaction between host and viral factors. Cell Mol. Immunol. 2021, 18, 539–555. [Google Scholar] [CrossRef]
- Crill, E.K.; Furr-Rogers, S.R.; Marriott, I. RIG-I is required for VSV-induced cytokine production by murine glia and acts in combination with DAI to initiate responses to HSV-1. Glia 2015, 63, 2168–2180. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Zheng, C. The Race between Host Antiviral Innate Immunity and the Immune Evasion Strategies of Herpes Simplex Virus 1. Microbiol. Mol. Biol. Rev. 2020, 84, e00099-20. [Google Scholar] [CrossRef]
- Zhao, J.; Qin, C.; Liu, Y.; Rao, Y.; Feng, P. Herpes Simplex Virus and Pattern Recognition Receptors: An Arms Race. Front. Immunol. 2020, 11, 613799. [Google Scholar] [CrossRef]
- Yu, H.; Bruneau, R.; Brennan, G.; Rothenburg, S. Battle Royale: Innate Recognition of Poxviruses and Viral Immune Evasion. Biomedicines 2021, 9, 765. [Google Scholar] [CrossRef]
- Chiang, J.J.; Sparrer, K.M.J.; Van Gent, M.; Lässig, C.; Huang, T.; Osterrieder, N.; Hopfner, K.-P.; Gack, M.U. Viral unmasking of cellular 5S rRNA pseudogene transcripts induces RIG-I-mediated immunity. Nat. Immunol. 2018, 19, 53–62. [Google Scholar] [CrossRef]
- Pomeranz, L.E.; Reynolds, A.E.; Hengartner, C.J. Molecular biology of pseudorabies virus: Impact on neurovirology and veterinary medicine. Microbiol. Mol. Biol. Rev. 2005, 69, 462–500. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Wang, M.; Cheng, A.; Yang, Q.; Wu, Y.; Jia, R.; Liu, M.; Zhu, D.; Chen, S.; Zhang, S.; et al. Innate Immune Evasion of Alphaherpesvirus Tegument Proteins. Front. Immunol. 2019, 10, 2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, J.; Wang, S.; Lin, R.; Mossman, K.L.; Zheng, C. Herpes simplex virus 1 tegument protein US11 downmodulates the RLR signaling pathway via direct interaction with RIG-I and MDA-5. J. Virol. 2012, 86, 3528–3540. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Zeng, Y.; Xu, S.; Chen, J.; Shen, G.; Yu, C.; Knipe, D.; Yuan, W.; Peng, J.; Xu, W.; et al. A Viral Deamidase Targets the Helicase Domain of RIG-I to Block RNA-Induced Activation. Cell Host Microbe 2016, 20, 770–784. [Google Scholar] [CrossRef] [Green Version]
- Kong, Z.; Yin, H.; Wang, F.; Liu, Z.; Luan, X.; Sun, L.; Liu, W.; Shang, Y. Pseudorabies virus tegument protein UL13 recruits RNF5 to inhibit STING-mediated antiviral immunity. PLoS Pathog. 2022, 18, e1010544. [Google Scholar] [CrossRef]
- Bo, Z.; Miao, Y.; Xi, R.; Zhong, Q.; Bao, C.; Chen, H.; Sun, L.; Qian, Y.; Jung, Y.S.; Dai, J. PRV UL13 inhibits cGAS-STING-mediated IFN-β production by phosphorylating IRF3. Vet. Res. 2020, 51, 118. [Google Scholar] [CrossRef]
- Zhu, Q.; Yu, T.; Gan, S.; Wang, Y.; Pei, Y.; Zhao, Q.; Pei, S.; Hao, S.; Yuan, J.; Xu, J.; et al. TRIM24 facilitates antiviral immunity through mediating K63-linked TRAF3 ubiquitination. J. Exp. Med. 2020, 217, e20192083. [Google Scholar] [CrossRef] [PubMed]
- Ning, F.; Li, X.; Yu, L.; Zhang, B.; Zhao, Y.; Liu, Y.; Zhao, B.; Shang, Y.; Hu, X. Hes1 attenuates type I IFN responses via VEGF-C and WDFY1. J. Exp. Med. 2019, 216, 1396–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Fu, Z.; Yin, H.; Han, Q.; Fan, W.; Wang, F.; Shang, Y. Macrophage Polarization Modulated by Porcine Circovirus Type 2 Facilitates Bacterial Coinfection. Front. Immunol. 2021, 12, 688294. [Google Scholar] [CrossRef]
- Lv, L.; Cao, M.; Bai, J.; Jin, L.; Wang, X.; Gao, Y.; Liu, X.; Jiang, P. PRV-encoded UL13 protein kinase acts as an antagonist of innate immunity by targeting IRF3-signaling pathways. Vet. Microbiol. 2020, 250, 108860. [Google Scholar] [CrossRef]
- Domingo-Calap, P.; Segredo-Otero, E.; Durán-Moreno, M.; Sanjuán, R. Social evolution of innate immunity evasion in a virus. Nat. Microbiol. 2019, 4, 1006–1013. [Google Scholar] [CrossRef]
- Ank, N.; West, H.; Bartholdy, C.; Eriksson, K.; Thomsen, A.R.; Paludan, S.R. Lambda interferon (IFN-lambda), a type III IFN, is induced by viruses and IFNs and displays potent antiviral activity against select virus infections in vivo. J. Virol. 2006, 80, 4501–4509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thaiss, C.A.; Zmora, N.; Levy, M.; Elinav, E. The microbiome and innate immunity. Nature 2016, 535, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.A.; Kagan, J.C. Toll-like receptors and the control of immunity. Cell 2020, 180, 1044–1066. [Google Scholar] [CrossRef]
- Ma, Z.; Ni, G.; Damania, B. Innate sensing of DNA virus genomes. Annu. Rev. Virol. 2018, 5, 341–362. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Beachboard, D.C.; Horner, S.M. Innate immune evasion strategies of DNA and RNA viruses. Curr. Opin. Microbiol. 2016, 32, 113–119. [Google Scholar] [CrossRef]
- Samanta, M.; Iwakiri, D.; Kanda, T.; Imaizumi, T.; Takada, K. EB virus-encoded RNAs are recognized by RIG-I and activate signaling to induce type I IFN. EMBO J. 2006, 25, 4207–4214. [Google Scholar] [CrossRef] [Green Version]
- Minamitani, T.; Iwakiri, D.; Takada, K. Adenovirus virus-associated RNAs induce type I interferon expression through a RIG-I-mediated pathway. J. Virol. 2011, 85, 4035–4040. [Google Scholar] [CrossRef] [Green Version]
- García-Sastre, A. Ten Strategies of Interferon Evasion by Viruses. Cell Host Microbe 2017, 22, 176–184. [Google Scholar] [CrossRef]
- Chan, Y.K.; Gack, M.U. Viral evasion of intracellular DNA and RNA sensing. Nat. Rev. Microbiol. 2016, 14, 360–373. [Google Scholar] [CrossRef]
- Yao, X.D.; Rosenthal, K.L. Herpes simplex virus type 2 virion host shutoff protein suppresses innate dsRNA antiviral pathways in human vaginal epithelial cells. J. Gen. Virol. 2011, 92 Pt 9, 1981–1993. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Yuzawa, E.; Imaizumi, T.; Matsumiya, T.; Yoshida, H.; Fukuhara, R.; Kimura, H.; Fukui, A.; Tanji, K.; Mori, F.; Wakabayashi, K.; et al. Retinoic acid-inducible gene-I is induced by interferon-gamma and regulates CXCL11 expression in HeLa cells. Life Sci. 2008, 82, 670–675. [Google Scholar] [CrossRef]
- Patel, J.R.; García-Sastre, A. Activation and regulation of pathogen sensor RIG-I. Cytokine Growth Factor Rev. 2014, 25, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.-Z.; Sarkar, D.; Emdad, L.; Barral, P.M.; Fisher, P.B. Central role of interferon regulatory factor-1 (IRF-1) in controlling retinoic acid inducible gene-I (RIG-I) expression. J. Cell Physiol. 2007, 213, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Guo, Z.; Kumar, A.; Wiens, M.; Gangappa, S.; Katz, J.M.; Cox, N.J.; Lal, R.B.; Sarkar, D.; Fisher, P.B.; et al. Influenza virus NS1-C/EBPβ gene regulatory complex inhibits RIG-I transcription. Antivir. Res. 2020, 176, 104747. [Google Scholar] [CrossRef] [PubMed]
- Koyanagi, N.; Kato, A.; Takeshima, K.; Maruzuru, Y.; Kozuka-Hata, H.; Oyama, M.; Arii, J.; Kawaguchi, Y. Regulation of herpes simplex virus 2 protein kinase UL13 by phosphorylation and its role in viral pathogenesis. J. Virol. 2018, 92, e00807-18. [Google Scholar] [CrossRef] [Green Version]
- Eaton, H.E.; Saffran, H.A.; Wu, F.W.; Quach, K.; Smiley, J.R. Herpes simplex virus protein kinases US3 and UL13 modulate VP11/12 phosphorylation, virion packaging, and phosphatidylinositol 3-kinase/Akt signaling activity. J. Virol. 2014, 88, 7379–7388. [Google Scholar] [CrossRef] [Green Version]
- Asai, R.; Ohno, T.; Kato, A.; Kawaguchi, Y. Identification of proteins directly phosphorylated by UL13 protein kinase from herpes simplex virus 1. Microbes Infect. 2007, 9, 1434–1438. [Google Scholar] [CrossRef]
- Tanaka, M.; Nishiyama, Y.; Sata, T.; Kawaguchi, Y. The role of protein kinase activity expressed by the UL13 gene of herpes simplex virus 1: The activity is not essential for optimal expression of UL41 and ICP0. Virology 2005, 341, 301–312. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Sequence (5′–3′) | Reverse Sequence (5′–3′) |
|---|---|---|
| Primer sequences for qPCR | ||
| hGAPDH | ATCAAGAAGGTGGTGAAGCA | GTCGCTGTTGAAGTCAGAGGA |
| hIFNB1 | GCACTGGCTGGAATGAGACT | CCTTGGCCTTCAGGTAATG |
| hISG56 | TTCGGAGAAAGGCATTAGA | TCCAGGGCTTCATTCATAT |
| hMX1 | AGCCACTGGACTGACGACTT | ACCACGGCTAACGGATAAG |
| hOASL | CCCTGGGGCCTTCTCTTC | TCCTAACAGTGCCATTCCCT |
| hTNF | AATAGGCTGTTCCCATGTAGC | AGAGGCTCAGCAATGAGTGA |
| hIL-6 | TAATGGGCATTCCTTCTTCT | TGTCCTAACGCTCATACTTTT |
| h-RIG-I | CTGGTTCCGTGGCTTTTTGG | CACCTGCCATCATCCCCTTA |
| h-RIG-I-Flag | GACTACAAGGACGACGATGA | AGTGTGGCAGCCTCCATTGG |
| h-MDA5 | AAAGCTCCTACCCGAGTGTG | GCTGCCCACTTAGAGAAGCA |
| h-MDA5-Flag | GACTACAAGGACGACGATGA | AGGCTCCACCTGGATGTACA |
| h-LGP2 | CAGCTGAGCCGACTTAGGAA | CGCAGCAGCAGTACTTAACC |
| pGAPDH | TACACTGAGGACCAGGTTGTG | TTGACGAAGTGGTCGTTGAG |
| pIFNB1 | TGCATCCTCCAAATCGCTCT | ATTGAGGAGTCCCAGGCAAC |
| pISG56 | TCCGACACGCAGTCAAGTTT | TGTAGCAAAGCCCTGTCTGG |
| pMX1 | GCTTTCAGATGCTTCGCAGG | TGTCGTATGGCTGATTGCCT |
| pOASL | CAGGCCAACAGGTTCAGACAG | CAGGAAACCGCAGACGATGT |
| pTNF | CGACTCAGTGCCGAGATCAA | CTCACAGGGCAATGATCCCA |
| pIL6 | AAGCTGCAGTCACAGAACGA | GGACGGCATCAATCTCAGGT |
| p-RIG-I | TCCTTCTGACTGCTAACGCT | ACTAAGGAAGGTGTCCAGCAG |
| p-MDA5 | TGAGGACTGATGTTTGATTCCAG | ACCTCTGCCCACCAAGATAGA |
| pLGP2 | CAGCCCTGCAAACAGTACGAC | CACTCCAGTTTCGGGTTCTC |
| Primer sequences for regular PCR | ||
| PRV UL13 | CCGGAATTCATGGACTACAAAGACGATG | CGCGGATCCTCAGGCATCGAGTTCG |
| hRIG-I-Luc | CGGGGTACCAAGTTTATCTGTAGGTTCAATG | GGAAGATCTGCCTCACTAGCTTTAAAGCC |
| hMDA5-Luc | CGGGGTACCCCAAGGTTTCATTTACTTCAAC | GGAAGATCTCCTGACTTTGGTTTCTGTTT |
| hLGP2-Luc | CGGGGTACCGGAGACCAGGTTTCCTTTCCAG | GGAAGATCTAGAAATGGAAACTGAAACTGAG |
| hRIG-I-Luc-M1 | ATTTGGACAACAGGTTATAAAGCTAAACAT | ATGTTTAGCTTTATAACCTGTTGTCCAAAT |
| hRIG-I-Luc-M2 | TTTGGACAACAGGTTATAAAGCTAAACAT | ATGTTTAGCTTTATAACCTGTTGTCCAAA |
| hMDA5-Luc-M1 | GCCTGGCGGGGATCAGGGAGACGC | GCGTCTCCCTGATCCCCGCCAGGC |
| hMDA5-Luc-M2 | AGCATGTGATTTAAAGGGGAAGTG | CACTTCCCCTTTAAATCACATGCT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, N.; Wang, F.; Kong, Z.; Shang, Y. Pseudorabies Virus Tegument Protein UL13 Suppresses RLR-Mediated Antiviral Innate Immunity through Regulating Receptor Transcription. Viruses 2022, 14, 1465. https://doi.org/10.3390/v14071465
Zhao N, Wang F, Kong Z, Shang Y. Pseudorabies Virus Tegument Protein UL13 Suppresses RLR-Mediated Antiviral Innate Immunity through Regulating Receptor Transcription. Viruses. 2022; 14(7):1465. https://doi.org/10.3390/v14071465
Chicago/Turabian StyleZhao, Ningning, Fan Wang, Zhengjie Kong, and Yingli Shang. 2022. "Pseudorabies Virus Tegument Protein UL13 Suppresses RLR-Mediated Antiviral Innate Immunity through Regulating Receptor Transcription" Viruses 14, no. 7: 1465. https://doi.org/10.3390/v14071465
APA StyleZhao, N., Wang, F., Kong, Z., & Shang, Y. (2022). Pseudorabies Virus Tegument Protein UL13 Suppresses RLR-Mediated Antiviral Innate Immunity through Regulating Receptor Transcription. Viruses, 14(7), 1465. https://doi.org/10.3390/v14071465