1. Introduction
Pseudomonas aeruginosa is an opportunistic pathogen known to cause various infections in humans, from mild, such as otitis externa, to life-threatening ones [
1]. As a human pathogen, it most often colonizes the epithelium of the lungs and urinary tract [
2]. Patients with burn wounds, AIDS, and cystic fibrosis (CF) are particularly at risk of developing serious
P. aeruginosa infections, which account for a high death rate in this population [
3,
4,
5]. Moreover,
P. aeruginosa has the ability to colonize medical instruments and is therefore one of the most common nosocomial pathogens.
This bacterium possesses a wide range of virulence factors that can be divided into those bound to the cell (e.g., lipopolysaccharides, flagella, pili) and those secreted from the cell (e.g., proteases, elastases, exotoxins, pyocyanin). However, the most important role in the pathogenicity of
P. aeruginosa is its ability to form biofilms. After binding to a surface,
P. aeruginosa undergoes a series of changes to adapt to a new way of life. Surface-bound cells grow and form microcolonies, while also producing an extracellular matrix. This extracellular matrix consists of exopolysaccharides, extracellular DNA (eDNA), and proteins, playing a role in biofilm adhesion and protection [
6,
7]. As the biofilm matures, the bacteria undergo physiological changes and become more resistant to environmental stresses. Due to its metabolic versatility, intrinsic and acquired antibiotic resistance, as well as biofilm formation and production of numerous virulence factors,
P. aeruginosa represents a serious global health concern.
Much has been completed to isolate
P. aeruginosa specific bacteriophages in order to use these viruses as alternative antimicrobial agents. The most studied bacteriophages are the tailed phages, and the reason behind this, is the ease of their isolation and multiplication. However, it is known that
P. aeruginosa can also be infected by filamentous phages (Pf). This particularly interesting but insufficiently examined phage group, belonging to the order
Tubulavirales, differs significantly from other bacteriophages in terms of morphology and life cycle [
8]. Filamentous phages have virions in which the major coat protein is helically organized around circular, positive-sense, single-stranded DNA ((+) ssDNA). These phages adhere to pili using CoaA protein, specific for a phage species, and upon penetration, ssDNA is converted to circular dsDNA designated as a replicative form (RF), which is used as a template for rolling-circle replication [
8]. After production of virion proteins, these phages extrude from cells, forming virions continuously, without causing host death. Besides CoaA protein, key proteins important for phage taxonomy are major coat protein CoaB and Zot protein that forms extrusion pores, and their amino-acid sequences are specific for a genus. The filamentous bacteriophages of
P. aeruginosa belong to the family
Inoviridae, and some of them are non-integrative, which replicate exclusively extrachromosomally, while others are integrative, being able to persist as prophages in a host genome [
9,
10]. The phages Pf1 and Pf3 are two previously described non-integrative filamentous phages of
P. aeruginosa, that currently are only
P. aeruginosa filamentous phages that fulfill criteria to be recognized as species by ICTV [
8]. Other
P. aeruginosa filamentous phages are known to be integrated into the genomes of their bacterial hosts: Pf4 and Pf5 persist in the form of prophages in strains PAO1 and UCBPP-PA14 (PA14), respectively [
11,
12,
13]. These two prophages actively produce new virions within their hosts. In addition, PfLES58 prophage from strain LESB58 and Pf7 prophage from strain PA7 are also
P. aeruginosa integrative filamentous phages, as well as many others in
P. aeruginosa genomes [
14], but they have been poorly examined.
Alongside Pseudomonas phage Pf1, the most widely studied is Pf4 phage. This phage influences PAO1 biofilm, particularly its formation and dispersal [
11,
13,
15], contributes to the appearance of small colony variants (SCVs) [
11], microcolony formation [
11], PAO1
in vivo virulence [
13], promotes PAO1 tolerance to tobramycin, but not to ciprofloxacin [
16], enhances resistance to 0.01% SDS [
13], decreases swimming [
17], and twitching motility [
18] etc. However, the data on Pf4 influence on other
P. aeruginosa strains still remains undetermined, although Pf4 specific genetic elements can be detected in more than 20% of all
P. aeruginosa strains [
14].
The aim of the study was to answer several clinically related questions—whether Pf4 can be induced during antibiotic or phage therapy, whether the released virions are capable to infect other P. aeruginosa strains, and how this phage influences virulence factors upon infection of new hosts. Additionally, based on the results, we proposed a new potential approach in phage therapy.
2. Materials and Methods
2.1. Pseudomonas aeruginosa Strains and Phages
Three strains of P. aeruginosa were used in the study: PAO1, LESB58, and UCBPP-PA14 (PA14), all naturally infected with filamentous (pro)phages: Pf4, PfLES58, and Pf5, respectively. The strain PAO1 was used to propagate phage Pf4 and check its induction, while other strains were used as hosts for Pf4 infection. Obligatory lytic phage JG024 was used to induce Pf4. JG024 phage was stored in SM buffer with 10% glycerol at −80 °C. Mueller Hinton (MH) broth (Torlak, Belgrade, Serbia) was used to store the strains, with 10% glycerol at −80 °C. The strains were propagated in MH broth at 37 °C for 24 h or on MH agar, to obtain colonies. Further subcultivation was performed using MH broth, MH agar, Luria Bertani (LB) agar with various percentage of agar, King’s A or King’s B medium, depending on experiment.
2.2. Antimicrobial Agents
In this study, discs of ciprofloxacin (CIP, 5 µg), gentamicin (GEN, 10 µg), tetracycline (TET, 30 µg), and streptomycin (STR, 300 µg) (Bioanalyse, Ankara, Turkey) were used. Additionally, the solutions of CIP, GEN, TET, STR, ceftazidime (CAZ), chloramphenicol (CHL), polymyxin B (PMB), and mitomycin C (MMC) (Sigma Aldrich, Saint Louis, MO, USA) were used for MIC determination.
2.3. Phage Pf4 Propagation
The strain PAO1 was inoculated in 600 mL LB broth and incubated at 37 °C for 24 h with shaking at 250 rpm. After incubation, the culture was centrifuged at 10,000×
g at 4 °C for 10 min to remove the bacteria [
19]. The phages were precipitated with 4% polyethylene glycol 6000 (PEG6000) (Sigma) and 0.5 M NaCl, and ultracentrifuged in CsCl equilibrium (0.375 CsCl g mL
−1) using Beckman Ti50 fixed angle rotor (133,000×
g, 4 °C, 42 h). After ultracentrifugation, the viral band was aspirated and dialyzed repeatedly in SM buffer (2 L of buffer per 1 mL of phage suspension).
2.4. Pf4 Induction from Original Host PAO1
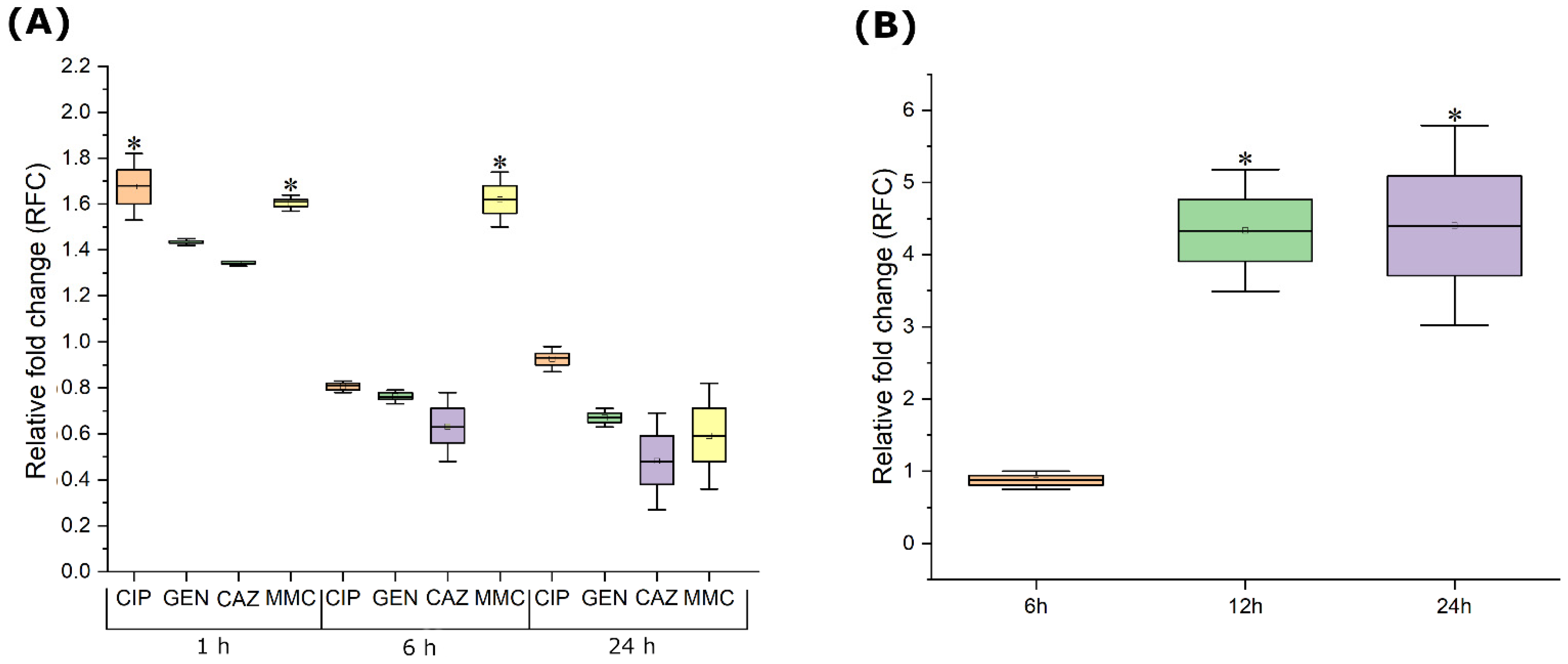
To determine the effect of subinhibitory concentrations of antimicrobials on Pf4 gene expression in PAO1 strain, 1/4 of obtained MIC values were used for all agents (CIP 0.0625 μg mL−1, GEN 0.5 μg mL−1, CAZ 16 μg mL−1, and MMC 0.5 μg mL−1). Similarly, PAO1 was treated with 0.25 MOI of JG024 phage (MIC equivalent 1 MOI).
In 50 mL of freshly prepared MH broth, 100 μL of the prepared bacterial suspension was added (0.5 McFarland density; ~108 CFU mL−1). The inoculated medium was incubated at 37 °C, 200 rpm, and the optical density of the contents was monitored continuously. After reaching the desired optical density of 0.5 McFarland density, one antimicrobial agent was added to each Erlenmeyer flask, and final volume in each flask was the same. The same volume of sterile distilled water was added in flasks used as negative controls. After 1 h, 6 h, and 24 h of incubation from treatment, samples were taken for bacterial RNA isolation.
In 50 mL of freshly prepared MH broth, 100 μL of the prepared bacterial suspension was added (0.5 McFarland density; ~108 CFU mL−1). The inoculated medium was incubated at 37 °C, 200 rpm, and the optical density of the contents was monitored continuously. After reaching the desired optical density of 0.5 McFarland density, the inoculated medium was treated with JG024. The same volume of SM buffer was added in a flask used as negative control. After 6 h, 12 h, and 24 h of incubation from treatment, samples were taken for bacterial RNA isolation.
The obtained treatment and control samples were immediately centrifuged at 12,000× g for 2 min. RNA was isolated using the GeneJET RNA Purification Kit (Thermo Scientific, Waltham, MA, USA). Concentrations of isolated RNA from all samples were uniformed to avoid variation in growth intensity and treated with DNAse I (1 U) for 30 min at 37 °C, followed by enzyme heat inactivation.
The isolated RNA was used for reverse transcription using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Bedford, MA, USA) with the following steps: 10 min at 25 °C, then 120 min at 37 °C, and finally 5 min at 85 °C. The reaction mixture was then cooled to 4 °C.
The newly designed primers Pf4Zot and Pf4CoaB (
Table S1) were used for the RT-qPCR in triplicate and in three independent repetitions, with the following cycling conditions: an initial cycle of 2 min at 50 °C, the next step at 95 °C for 10 min, then a step at 60 °C for 1 min, followed by a step at 55 °C for 5 s, and the final step lasted 5 min at 95 °C. The Ct values of each sample represent the average + S.E. of the results of replicates. No template controls served as negative controls, and Pf4 DNA served as a positive control. To normalize data, two primer pairs targeting two
P. aeruginosa housekeeping genes for
rpoD and
proC were used (
Table S1). Transcriptional changes were calculated using the 2
−ΔΔCT method and changes in relative gene expression ≥1.5 or ≤0.67 were considered significant [
20].
2.5. Pf4 Phage Infection of Other P. aeruginosa Strains
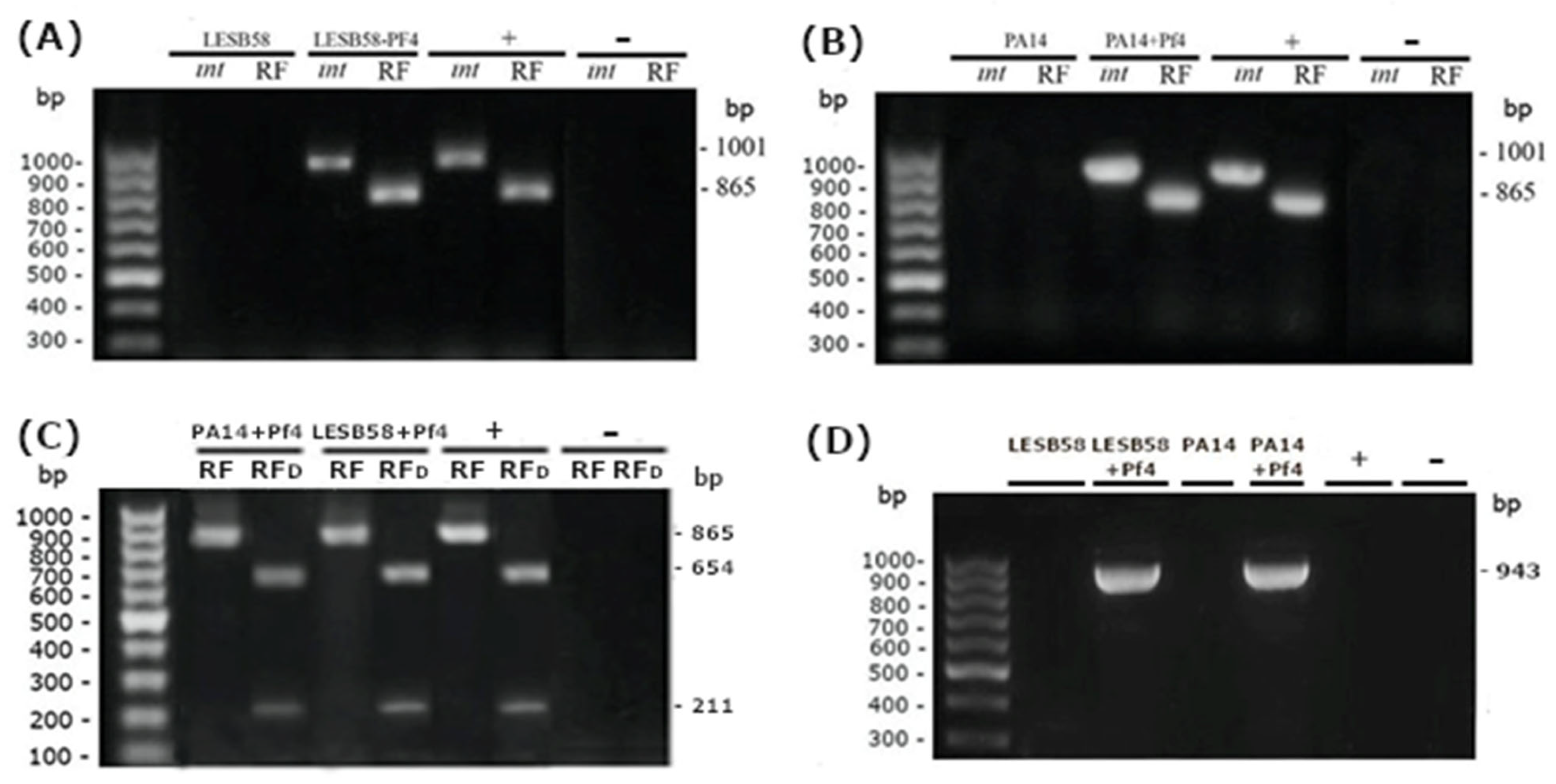
In order to achieve Pf4 infection of LESB58 and PA14, bacteria and phage were mixed at MOI = 10 and incubated overnight at 37 °C. After incubation, the bacteria were centrifuged at 6000× g for 2 min. The bacterial pellet was resuspended in PBS buffer and centrifuged again. This step was repeated one more time, to remove unadhered Pf4 virions from the supernatant. Over bacterial pellet, 1 mL of MH broth was added and the bacteria were incubated overnight at 37 °C. Re-washing the bacterial cells and adding freshly prepared medium was repeated twice, to completely remove Pf4 that did not infect bacterial cells, but potentially remained in the medium. The bacteria were then inoculated on MH agar at 37 °C. The subcultivation was repeated three more times in order to obtain stable lysogens.
Upon infection, the presence of Pf4 (pro)phages in different
P. aeruginosa strains was determined by PCR. Several colonies from each presumed lysogenic strain were used as samples, in such a way that one half of the colony remained on agar in case of successful confirmation of superinfection. Two pairs of primers were used to determine the presence of replicative form (RF) and gene for Pf4 integrase (
intF4) (
Table S1). Thermal cycling conditions for both primer pairs were as follows: an initial cycle of 94 °C for 5 min followed by 35 cycles of 94 °C for 30 s, annealing at 55 °C for 20 s, and extension at 72 °C for 60 s, with a final 7 min extension at 72 °C. PCR products were analyzed by 1.5% agarose gel with ethidium bromide. PAO1 DNA was used as a positive control and sterile distilled water was used as a negative control.
To further verify the presence of Pf4 (pro)phage in the infected P. aeruginosa strains, the obtained PCR products for the RF were cut using enzyme HpaII (FastDigest, Thermo Fisher Scientific, Vilnius, Lithuania). For digestion, 10 μL of PCR product (~0.2 μg), 1 μL of HpaII enzyme, 10 μL of FastDigest Green buffer, and 17 μL of dH2O were mixed. The FastDigest mixture was then incubated for 5 min at 37 °C. At the end of the incubation, the restricted PCR products were separated using 1% agarose gel with ethidium bromide. According to in silico analyses, HpaII cut the product of Pf4 RF, giving fragments of 654 and 211 bp.
All gels were documented using BioDocAnalyze Transiluminator (Biometra, Gottingen, Germany).
For confirmation of prophage formation, we postulated that Pf4 was integrated into PA14 and LESB58 using the same
att site in bacterial genome. We in silico included the phage DNA into bacterial genome and designed primer pairs that comprise both bacterial and viral DNA (
Table S1). For purification of PCR products, ExoSAP Master Mix was prepared (100 μL of Exonuclease I (20 U/μL), 200 μL of FastAP (1 U/ μL), 60 μL of Exonuclease I Reaction Buffer 10×, and 240 μL of PCR water). In 5 μL of PCR reaction, 1 μL of ExoSAP Master Mix was added and the samples were incubated for 15 min at 37 °C. Enzyme inactivation was performed at 85 °C for 15 min. For the sequencing purposes, 5 μL of purified PCR product was mixed with 5 μL of forward or reverse primer at a concentration of 5 μM. Sequencing of PCR products was conducted using capillary electrophoresis on an ABI 3730 × l Genetic Analyzer (Applied Biosystems). The alignment was conducted by using DNADynamo program with final manual re-checking. The length of analyzed sequences was 943 bp for both lysogenic strains, LESB58 + Pf4 and PA14 + Pf4.
2.6. Production of Pf4 and Indigenous Phages in Superinfected Strains
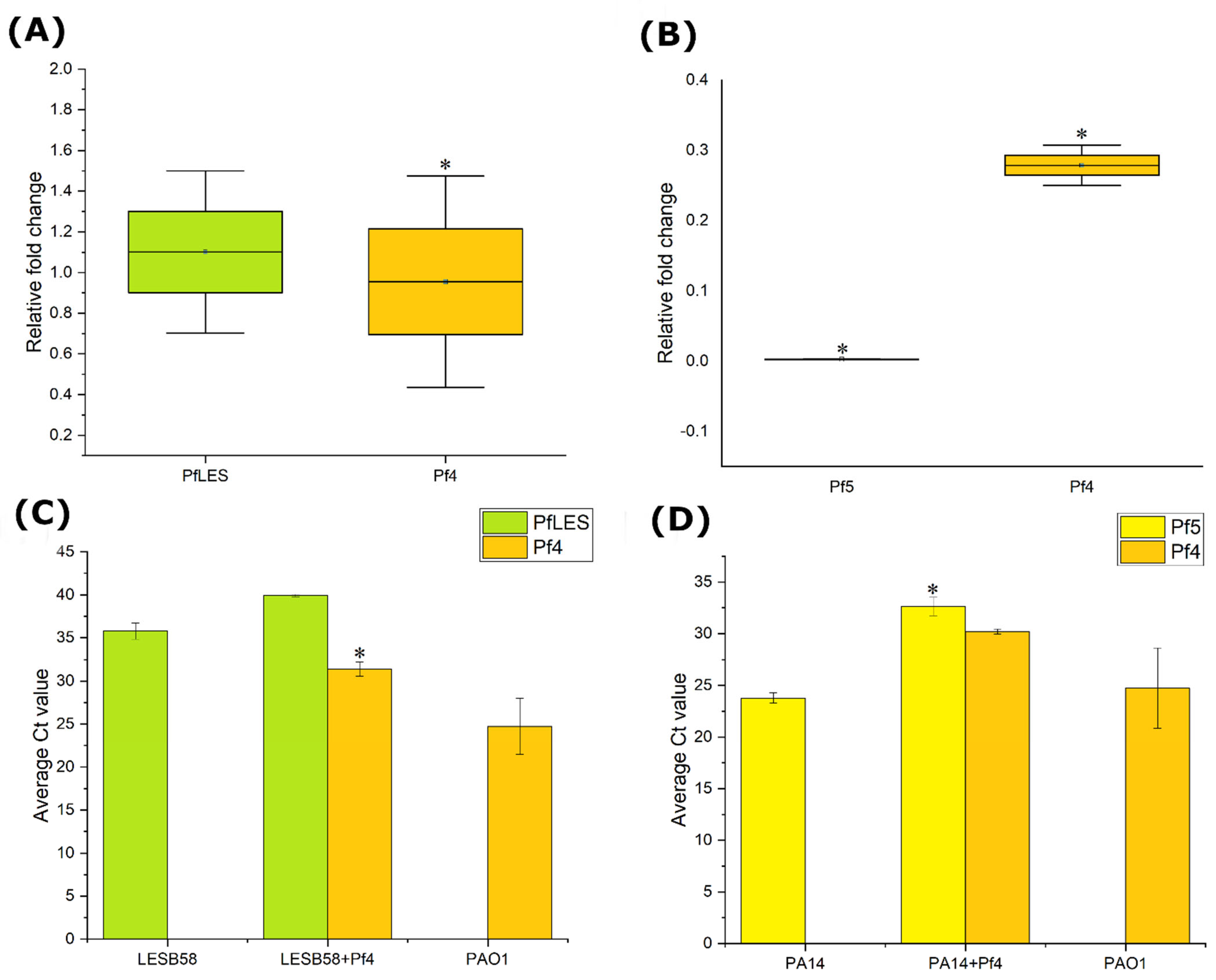
To confirm the production of Pf4 phage in new hosts of
P. aeruginosa, lysogenic and non-lysogenic strains were inoculated in 20 mL of MH broth with a final abundance of ~10
6 CFU mL
−1. All strains were incubated for 12 h, at 37 °C, at 200 rpm. After 6 h of incubation, 1 mL of bacterial suspensions were used to isolate RNA using GeneJET RNA Purification Kit (Thermo Scientific, USA). The resulting RNA was equalized and purified with 1 μL DNAse I (1 U) for 30 min at 37 °C followed by DNAse inactivation with 1 μL of 50 mM EDTA at 65 °C for 10 min. The purified RNA was translated into cDNA using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Bedford, MA, USA) with the following steps: 10 min at 25 °C, then 120 min at 37 °C, and finally 5 min at 85 °C. The reaction mixture was then cooled to 4 °C. The cDNA of all samples was double diluted and then RT-qPCR was used to check the expression of Pf4 phages in lysogenic hosts, as well as the expression of Pf phages that naturally infect given hosts. The primers Pf4CoaA, PfLES58CoaA, and Pf5CoaA (
Table S1), specific enough to discriminate all three phages, were designed using PrimerBlast (NCBI) and Oligoanalyzer. To normalize data, two primer pairs targeting two
P. aeruginosa housekeeping genes were used (
rpoD and
proC).
Furthermore, after 12 h of incubation, 10 mL of the bacterial suspensions were purified by centrifugation (6000×
g, 10 min) followed by filtration through 0.22 μm pores. PEG6000 and NaCl were added to the purified suspensions with final concentrations of 4% and 0.5 M, respectively, and left to incubate overnight at 4 °C. Prior to isolating DNA from the precipitated phages, residual DNA that is not from intact virions was removed by DNase I [
21]. Five units of DNase I (≥2500 units mL
−1) were added to 200 μL of each sample, and then incubated at 37 °C for 10 min. To isolate DNA from phage particles without the need for additional purification steps, DNase I pre-treated phage samples were heat-denatured at 100 °C for 15 min. The viral DNA was then 100-fold diluted and then used for qPCR to compare the production of Pf4 with other Pf phages in newly infected strains. The qPCR primer pairs for Pf4, PfLES58, and Pf5 RF were designed (
Table S1).
The qPCR was performed in triplicates and in three independent repetitions, with the following cycling conditions: an initial cycle of 2 min at 50 °C, the next step at 95 °C for 10 min, then a step at 60 °C for 1 min, followed by a step at 55 °C for 5 s, and the final step lasted 5 min at 95 °C. The Ct values of each sample represent the average + S.E. of the results of replicates. No template controls served as negative controls.
2.7. Pf4 Influence on Phenotype of Alternative Hosts
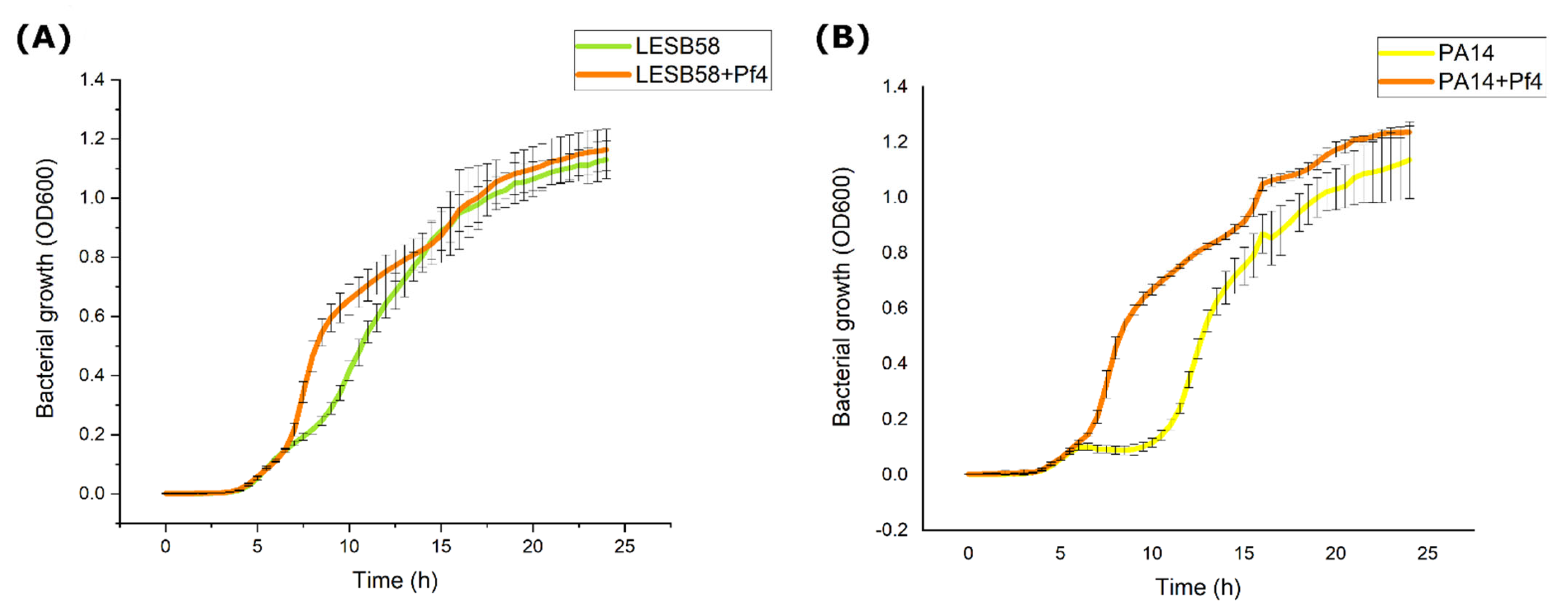
2.7.1. Growth Kinetics
Prepared bacterial suspensions of 0.5 McFarland density (~108 CFU mL−1) of overnight cultures were diluted in PBS buffer at a ratio of 1:100 (v/v) and then further diluted in a liquid double concentrated MH broth in a ratio of 1:1 (v/v). Then, 200 μL of inoculated medium was added to the wells of a microtiter plate in triplicates. The microtiter plates were incubated for 24 h at 37 °C in a spectrophotometer with continuous shaking (Thermo Scientific™ Multiskan™ GO) and the absorbance was measured at 600 nm every 30 min. The obtained average values for lysogenic strains were compared with non-lysogenic, original strains. The experiment was conducted in triplicates and results were averaged.
2.7.2. Autoaggregation
The autoaggregation test was performed by the method of Basson et al., (2007) [
22]. The overnight bacterial cultures were aliquoted 1 mL each into sterile tubes and centrifuged for 5 min at 6000×
g. The resulting supernatant was removed and the cells were resuspended in 1 mL of PBS. These steps were repeated twice. The washed cells were used for the preparation of two series of bacterial suspension. The first series was left untreated containing 800 µL of PBS and 200 µL of resuspended washed bacterial cells, and was prepared prior to measurements. The second series also contained 800 µL of PBS and 200 µL of resuspended washed bacterial cells but it was incubated for 1 h at 37 °C. At the end of incubation, the second series was centrifuged for 2 min at 650×
g. The resulting supernatant was aliquoted into a microtiter plate in three replicates of 200 µL each. The optical density of aliquoted series was read on a spectrophotometer (Thermo Scientific™ Multiskan™ GO) at 650 nm and the degree of autoaggregation was further determined. The percentage of autoaggregation (%) of the test isolates was determined using the following equation: [(OD0 − OD60)/OD0] × 100, in which OD0 refers to the initial optical density of strains (first series), while OD60 means the optical density obtained after 60 min of treatment (second series). Depending on the degree of autoaggregation, bacterial strains were characterized as highly aggregative (>50%), moderately aggregative (20–50%), and non-aggregative (<20%) [
23]. The obtained average values for lysogenic strains were compared with non-lysogenic, original strains. The experiment was performed in at least three independent triplicates.
2.7.3. Cell Surface Hydrophobicity
Bacterial Adherence to Hydrocarbons (BATH) was used to determine the hydrophobicity of the lysogenic strains [
24]. The overnight bacterial cultures (1 mL) were aliquoted into sterile tubes and centrifuged for 2 min at 6000×
g. The resulting supernatant was removed, and the cells were resuspended in 1 mL of PUM buffer (K
2HPO
4 + 4H
2O 22.2 g, KH
2PO
4 + 4H
2O 7.26 g, urea 1.8 g, MgSO
4 + 7H
2O 0.2 g, dH
2O 1 L). These steps were repeated twice. The washed cells were used to prepare two series of bacterial suspensions. The first series was untreated, containing 900 µL of PUM buffer and 200 µL of resuspended washed bacterial cells. The second series contained 2.7 mL of PUM buffer and 600 µL of resuspended washed bacterial cells (bacterial count was 2 × 10
8 CFU mL
−1). The second series was amended with 400 μL of hydrocarbon n-hexane. The tubes were vortexed for 2 min and then left for 15 min at room temperature to separate the phases. The aqueous phase was transferred to new tubes and left in the refrigerator overnight for the hydrocarbons to evaporate. The samples were aliquoted into a microtiter plate in three replicates of 200 µL each. The optical density was read on a spectrophotometer (Thermo Scientific™ Multiskan™ GO) at 560 nm and the degree of hydrophobicity was determined. The percentage of hydrophobicity (%) was determined using the following equation: [(A0 − A1)/A0] × 100, in which (A1) are optical densities of the treated suspensions compared to the suspensions used as a control (A0). Depending on the degree of adhesion to the hydrocarbon, the strains were characterized as extremely hydrophobic (>50%), moderately hydrophobic (20–50%), and hydrophilic (<20%) [
24]. The obtained average values for lysogenic strains were compared with non-lysogenic, original strains. The experiment was performed in at least three independent triplicates.
2.7.4. Motility Tests
Twitching, swarming, and swimming motility were assessed by stabbing P. aeruginosa strains into 1.5%, 0.7%, and 0.3% agar plates, respectively. After 24 h of incubation at 37 °C, 1.5% and 0.7% LB agar medium, for the determination of twitching and swarming motility of lysogenic and non-lysogenic strains, was removed. Then, to the inside of each Petri dish, 0.4% crystal violet solution was added. In this way, a better visualization of the movement of lysogenic and non-lysogenic strains was achieved. Swimming motility was determined on 0.3% agar plate surface. The experiment was performed in at least three independent triplicates.
2.7.5. Biofilm Formation on Polystyrene Surface
The biofilm formation by
P. aeruginosa was determined by the method of Knezevic et al., (2008) [
25]. The overnight bacterial cultures were aliquoted into sterile cuvettes and centrifuged for 2 min at 6000×
g. The resulting supernatant was removed and the cells were resuspended in 1 mL of PBS buffer. These steps were repeated twice. The bacterial suspensions of 0.5 McFarland density (~10
8 CFU mL
−1) were made. Suspensions were diluted in PBS buffer at a ratio of 1:100 (
v/
v) and then further diluted in MH broth in a ratio of 1:1 (
v/
v). Then 300 μL of inoculated medium was added to the wells of a microtiter plate. The microtiter plates were incubated for 24 h at 37 °C. After incubation, the medium was carefully removed using a multichannel pipette. The washing of microtiter plate wells was performed twice by 300 µL of PBS. The fixation of the formed biofilm was achieved by adding 300 µL of absolute methanol for 15 min. The methanol was removed and the plate was allowed to dry for 15 min at 44 °C. After that, biofilm was stained by 0.4% crystal violet for 15 min. Afterwards, the microtiter plate was submerged in tap water to remove excess crystal violet. After drying the plate, the dye was dissolved by adding 300 µL of 33% acetic acid. The microtiter plate was left at room temperature for 20 min. The optical density for each well was measured at 595 nm on a spectrophotometer (Thermo Scientific™ Multiskan™ GO). Assessment of cell adhesion was determined using ODc value, which represents the mean of the optical density of the negative control (OD) for the microtiter plate tested, which is increased by three standard deviations obtained for the negative controls.
P. aeruginosa strains were classified according to the following criteria: OD ≤ ODc = unadherent; ODc < OD ≤ (2 × ODc) = poorly adherent; (2 × ODc) < OD ≤ (4 × ODc) = moderately adherent (4 × ODc) < OD = highly adherent [
26]. The obtained average values for lysogenic strains were compared with non-lysogenic, original strains. This test was conducted in triplicates in three independent repetitions.
2.7.6. Pyocyanin Production
Quantification of produced pyocyanin was determined by using a modified method by Essar et al., (1990) [
27]. Bacterial suspensions with an optical density of 0.5 McFarland (~10
8 CFU mL
−1) were made from overnight cultures and then further diluted 1:100 (
v/
v). To 10 mL of King’s A broth, 100 µL of each strain was added and incubated at 37 °C for 24 h at 200 rpm. After incubation, samples were centrifuged at 6000×
g for 10 min, and resulting supernatants were collected. To 7.5 mL of supernatant, 4.5 mL of chloroform was added, vortexed 2 × 10 s, and the samples were centrifuged at 4000×
g for 10 min. Only 3 mL of the resulting blue layer at the bottom was transferred to a new tube. To each tube, 1.5 mL of 0.2 M HCl was added and then vortexed 2 × 10 s. At this step, the blue color turned into pink (top layer). Samples were centrifuged again at 4000×
g, 10 min, and 1 mL from the pink layer was transferred to a new tube. Spectrophotometric measurements were performed in microplates at 520 nm and pyocyanin concentration (μL mL
−1) was calculated by multiplying the obtained values and constant 17.072. As a blank, 0.2 M HCl was used. The experiment was performed in triplicates and at least on three independent occasions.
2.7.7. Pyoverdine Production
Quantification of pyoverdine was determined using a modified method by Déziel et al., (1991) [
28]. Bacterial suspensions with an optical density of 0.5 McFarland (~10
8 CFU mL
−1) were made from overnight cultures and then further diluted 1:100. To 2 mL of King’s B Medium, 100 µL of each strain was added and incubated at 37 °C for 24 h at 200 rpm. At the end of the incubation, 1 mL was used to determine the total number of bacteria. The residual overnight cultures were centrifuged and the resulting supernatants were used to quantify the relative concentration of pyoverdine by measuring the fluorescence (Fluoroscan Ascent FL, Thermo Labsystems, Waltham, MA, USA) at 460 nm after excitation with wavelength 340 nm. The experiment was performed in triplicates and at least on three independent occasions.
2.7.8. SCV Production
Non-lysogenic and Pf4 lysogenic strains were inoculated into 5 mL of freshly prepared Luria–Bertani broth and incubated at 37 °C for 5 days without agitation. At the end of the incubation, the bacterial suspensions were centrifuged (6000×
g, 5 min) and the resulting pellet was resuspended in 2 mL of PBS. This washing step was repeated once more, and then serial dilutions were made. The total numbers of wild-type and SCVs’ colonies for each strain were determined by the spread plate method on Luria–Bertani agar with 0.04% Congo Red [
29]. Colony counting was performed after 2 days of incubation at 37 °C. The experiment was conducted in triplicates and results were averaged.
2.8. Antibiotic Susceptibility
For preliminary estimation of antibiotic sensitivity of uninfected and Pf4-infected
P. aeruginosa strains, a disk diffusion method was applied using discs of CIP, GEN, TET, and STR [
30].
The antibiotic susceptibility of lysogenic strains was further determined by determination of minimal inhibitory concentration (MIC). Prepared bacterial suspensions of 0.5 McFarland density (~10
8 CFU mL
−1) of overnight cultures were diluted in MH broth (1:100
v/
v). Then 100 μL of inoculated medium was added to the wells of a microtiter plate. The same volume of antibiotics was added to the wells, with the final concentration of each antibiotic in the microtiter plates ranging from 0.0625 to 128 μg mL
−1. The following antibiotics were used: CIP, GEN, STR, TET, CAZ, CHL, and PMB. Total bacterial counts in each well of the microtiter plate were ~1 × 10
6 CFU mL
−1. The microtiter plates were incubated for 18 h at 37 °C, after which 10 μL of 0.1% solution of 2,3,5-triphenyltetrazolium chloride (TTC) was added to each well of the plate, which was reduced to red formazan by the dehydrogenases of viable bacterial cells. The microtiter plates were further incubated for 2 h at 37 °C, after which the MIC value for each antibiotic was read. The lowest concentration of antibiotics needed to prevent coloring of TTC to red formazan was considered as an MIC value. The reference strain
Escherichia coli ATCC 25922 was used as a control. The experiment was performed in at least three independent triplicates. The obtained results are presented as geometric mean. The reduction of MIC after phage infection by one value was not taken as statistically significant. Lysogenic and non-lysogenic strains have been characterized as sensitive, intermediate sensitive or resistant to antibiotics, tested according to the recommended criteria for
P. aeruginosa or, if not available, for other non-enterobacteriace (for TET, STR, and CHL) [
31].
2.9. Statistics
To compare virulence factors of non-lysogenic and lysogenic strains, Statistica 7.1 software was used. Normality of data distribution was determined by Kolmogorov–Smirnov Test of Normality. Difference of normally distributed data were tested by parametric paired t-test, while data without normal distribution were analyzed by Wilcoxon signed rank test. Confidence interval was CI95, and p values <0.5 were considered statistically significant.
4. Discussion
One the best known P. aeruginosa strains is PAO1, which harbors an integrative filamentous phage Pf4. Its influence on original host phenotypic properties is well established. Here, we examined inducibility of this phage, its possibility to infect other P. aeruginosa strains, and Pf4 contribution to their virulence factors.
The phage Pf4 can be produced in greater amounts, approx. more than 50%, after exposure to subinhibitory concentrations of ciprofloxacin and mitomycin C. While ciprofloxacin increased Pf4 phage production only briefly after PAO1 exposure to subinhibitory concentrations, mitomycin C had prolonged activity up to 6 h. Since both antimicrobials triggered the SOS response [
32,
33], they probably affected Pf4 production by the same mechanism. The reason why this induction is only contemporary may be related to the bacterial adaption to a stressful environment and SOS response termination [
32]. Mitomycin C has been previously used to test the induction of Pf4 phages of PAO1 strain, and it was found that the treated strain released superinfective Pf4 forms, determined by plaque assay, after the use of mitomycin C in concentrations higher than 10 μg mL
−1 [
15]. Accordingly, both ours and previous results indicated that Pf4 belongs to the group of mitomycin C inducible phages. On the other hand, gentamicin and ceftriaxone were not able to enhance Pf4 production and they were unable to activate SOS response, so this additionally supports the assumption related to SOS response. Moreover, for the first time, we proved that the infection of PAO1 with obligate lytic phage JG024 at MOI 0.25 can affect the expression of Pf4 prophage key genes up to 24 h after infection. Namely, this MOI could not cause complete lysis of bacteria during 24 h, probably due to the appearance of phage- resistant mutants. Thus, in such a system, phage Pf4 production is enhanced up to four times, probably as a result of altered metabolism of the cells and Pf4 regulatory mechanisms in phage-resistant, i.e., mutated cells. The results indicate that ciprofloxacin, mitomycin C, and obligatory lytic phages can induce filamentous phages during therapy and that they should be applied with precautions.
Strains of
P. aeruginosa LESB58 and PA14 are naturally infected with filamentous phages PfLES58 and Pf5, respectively, and can successfully be superinfected with Pf4 phages. The presence of Pf4 RF in these strains, as well as in the isolated viral DNA, indicates that this phage established a chronic productive infection in the given strains. Alongside the persistence of phage Pf4 in the RF, i.e., extrachromosomally, the sequencing results indicate that Pf4 was integrated in the genome of PA14 and LESB58 as a prophage in the proximity of tRNA-Gly, as in PAO1. The successful infection of the strains by Pf4 is not surprising, as it has been previously documented that one bacterial strain can carry several different or even the same prophages from the family
Inoviridae [
14]. Since all
Inoviridae utilize pili as receptors, it is expected that piliated cells, including PA14 and LESB58, are prone to infection by filamentous phages. The superinfection exclusion was not observed between PfLES58 or Pf5 and phage Pf4. PfLES58 phage expression in the presence of Pf4 phage was not significantly altered, while in the case of Pf5 phage, the change was drastic—Pf4 phage significantly reduced the expression and production of Pf5 virions in the PA14 + Pf4 strain. Pf4 phage expression in the first 6 h of incubation was also statistically significantly reduced. However, viral DNA analyses indicated that Pf4 phages reached a similar number of virions relative to PAO1, while Pf5 phage production remained reduced. In LESB58 + Pf4, the expression of both phages was unchanged, but PfLES58 predominated in virion production, and there was a statistically significant decrease in Pf4 virion in this lysogenic strain. This indicates that Pf4 virions, released increasingly during therapy, can successfully infect new strains of
P. aeruginosa, establishing very complex interactions with other indigenous filamentous (pro)phages.
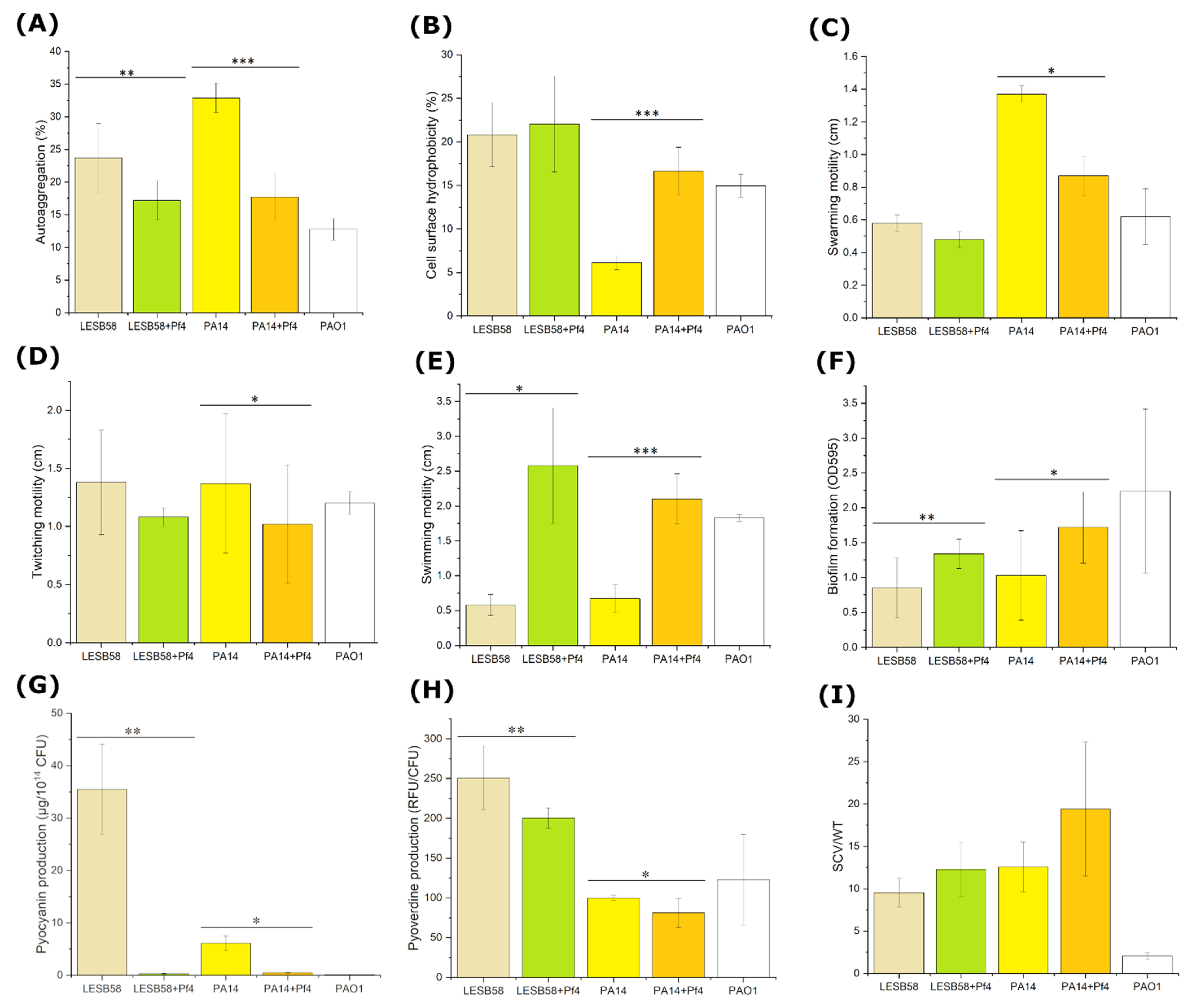
The phage Pf4 influence on PAO1 phenotype is well documented in the literature, and here it was confirmed that it also changed virulence factors of alternative hosts PA14 and LESB58.
Filamentous phages usually reduce bacterial growth, as reported for
Xanthomonas citri [
34,
35],
Xanthomonas campestris [
36],
Ralstonia solanacearum [
37],
Pseudoalteromonas [
38],
Vibrio alginolyticus [
39], and
E. coli [
40,
41]. However, there are known cases where filamentous phages do not affect the growth of their host as in
X. campestris [
42],
R. solanacearum [
43], and
Yersinia pestis [
44]. Here, we showed for the first time, that filamentous phages can slightly increase bacterial growth, but only contemporarily. The potential explanation is related to the fact that Pf4 encoded a toxin-antitoxin system type II, having an antitoxin with conserved sequence Phd_YeFM [
45]. This antitoxin binds to their toxin partners, can bind DNA via the N-terminus and repress the expression of operons containing genes encoding the toxin and the antitoxin. This domain complexes with Txe toxins with various domains, and is present in
P. aeruginosa cells [
46], which can partially explain the slight stimulation of growth. After 24 h incubation, the difference in growth cannot be observed, so the Pf4 infection has no long-term influence on PA14 and LESB58 growth.
The ability of cells to aggregate can play an important role in the initial phases of biofilm formation. Addy et al., (2012) [
47] hypothesized that the presence of filamentous phages on the surface of a bacterial cell may contribute to changes in the nature of the cell membrane, and increase cell to cell interactions which would contribute to high local cell densities. However, it seems the opposite happened with PA14 and LESB58
P. aeruginosa cells upon infection with Pf4 phage, as autoaggregation was decreased—non-lysogenic strains were moderately aggregative, but after infection with Pf4 phage, they became non-aggregative.
Upon the phage Pf4 infection, a slight increase in hydrophobicity in both examined lysogenic strains was observed, similar to findings for Ralstonia phage φRSS1 [
47]. However, the lysogenic strains remained either hydrophilic or moderately hydrophobic, i.e., without qualitative change in hydrophobicity. The increase in hydrophobicity can be explained by the fact that upon filamentous phage infection, lipids of the outer membrane are altered with change of the relative concentration of phospholipids [
48].
Several cell surface-associated structures, such as flagella and type IV pili, are essential for adhesion and microcolony formation, respectively [
49,
50]. Flagella act to overcome repulsive forces between the bacterium and the surface to allow the initial contact [
51]. This is the first report on increased swimming motility upon a filamentous phage infection that correlates with increased biofilm production, since bacterial motility plays an important role in the initial stages of biofilm formation [
52]. This finding is very interesting, since difference in swimming motility was not previously established for PAO1 strain infected with Pf4 and deprived of the prophage [
17]. Similar lack on swimming motility influence was observed for other phages, such as phage SW1 of
Shewanella piezotolerans [
53]. On the other hand,
Xanthomonas cells infected with XacF1 showed reduced swimming activity [
34]. As this is the first report on swimming motility increase upon filamentous phage infection, this phenomenon deserves further elucidation.
Both swarming and swimming motility are a flagellum-dependent form of movement observed in
P. aeruginosa. Overlapping sets of regulators have been found to affect swarming motility and biofilm formation in a reciprocal manner [
54,
55]. For example, strains lacking GacA, responsible for biofilm formation and EPS production, had increased swarming motility [
56], while strains with increased EPS production had reduced swarming motility [
54]. The presence of Pf4 in
P. aeruginosa strains led to a decrease in swarming motility in both strains, with significant difference observed for Pf4-infected PA14 strain. A similar example of decreased swarming motility caused by filamentous phages can be found in
Erwinia amylovora infected with PEar filamentous phages [
57], and
S. piezotolerans infected with SW1 phage [
53]. Together, these results confirm that the presence of filamentous phages may reduce swarming motility in different bacterial species, probably by change in the expression of key genes responsible for this type of motility.
Using type IV pili, bacteria exhibit twitching motility, which is movement over solid surfaces [
58]. The twitching motility is considered an important bacterial property that leads to grouping of cells into microcolonies [
49]. Furthermore, type IV pili are bacterial adhesins significant for bacterial interaction with mammalian cells, but also for autoaggregation. As indicated, filamentous phages use these structures as receptors, when establishing infection of bacteria [
59]. The results of the present study showed that in the presence of Pf4, there was a slight decrease in twitching motility in both lysogenic strains, with statistically significant difference observed for PA14 strain. This is not the first report regarding Pf phages and reduction of twitching motility. Secor et al., (2017) [
18] demonstrated that PAO1 strain superinfected with Pf4 phage exhibited minimal twitching motility. Similarly, filamentous phages of other bacterial species also reduced twitching motility, such as Xanthomonas phage XacF1 [
34], Ralstonia phage RSS51 [
43], and Ralstonia phage RSM3 [
60]. The strains used in this study, being naturally infected with other types of filamentous phages, produced Pf4 phage in addition, so the numerous phages can reinfect cells adsorbing to pili and impair their function. Another possibility is that Pf4, directly or indirectly, shuts down genes for pili production, to prevent superinfection by other similar phages, which should be further examined. The lysogenic strains with Pf phages are thought to be less invasive due to impaired twitching motility and to have a phenotype similar to twitching-deficient non-invasive
P. aeruginosa phenotypes [
61]. Finally, this finding is in correlation with the detected decrease in autoaggregation.
Bacteria often switch from a free-living lifestyle to a surface-adapted multicellular lifestyle known as a biofilm. It has been estimated that 60% of all
P. aeruginosa infections are biofilm related [
6,
62], and there is substantial evidence that biofilm plays an important role in persistent infections, e.g., lungs in CF patients. Our results show that
P. aeruginosa Pf4 lysogenic strains formed more quantity of biofilm compared to their non-lysogenic counterparts. For PAO1 strain, it is well documented that Pf4 phage plays a crucial role in biofilm formation and persistence [
11,
13,
16]. Similar to type IV pili, which facilitate bridging and permanent attachment of cells to the surface, the presence of a larger number of filamentous phages on the surface of the bacterial cell could have the same impact on biofilm formation [
11,
63], contributing to microcolony expansion [
13]. However, this cannot simply explain our results, as autoaggregation was decreased in Pf4-infected strains, and the same pertains to twitching motility, both considered important to biofilm formation. The enhanced cell surface hydrophobicity and swimming motility also contributed to cells’ better interaction with surface, and the established biofilms were probably further more intensively developed, as numerous phage particles contributed to the biofilm matrix structure. The role of Pf4 in biofilm matrix structure [
13,
16], as well as in biofilm volume and thickness [
64] were previously documented for PAO1 strain. Cell lysis by Pf4 phage may additionally contribute to better biofilm production, and this phenomenon was confirmed in the original host PAO1 [
15]. The cell lysis released nutrients into the biofilm matrix [
65,
66], while released eDNA contributed to matrix structure [
67]. Accordingly, the strains infected with Pf4 phage produced more quantities of biofilm than uninfected counterparts, probably not because of enhanced initial adhesion and more efficient microcolony formation, but rather due to a more complex biofilm matrix architecture which occurred in later phases of biofilm development.
Pyocyanin is a human cytotoxin, which is considered to be an extremely important secreted virulence factor of
P. aeruginosa [
68]. Infection by Pf4 phages severely inhibited pyocyanin production of lysogenic strains. Ismail et al., (2021) [
69] demonstrated that PAO1 without Pf4 phage produced less pyocyanin compared to wild-type after 24 h of incubation. Although it seemed that Pf4 stimulated pyocyanin production in its original host, it decreased pyocyanin production in PA14 and LESB58
P. aeruginosa strains. Pyoverdine is a water soluble siderophore, i.e., iron-chelating molecule, able to trigger activation of virulence factors, exotoxin A and protease PrpL, when it is in the form of ferripyoverdine [
70]. A significant decrease in pyoverdine production was observed in both lysogenic strains. Similarly, the PAO1 mutant in the absence of Pf4 prophage was shown to produce significantly more pyoverdine than its wild-type [
69]. The results obtained for
P. aeruginosa exopigment production suggest that the Pf4 decreased toxicity of the examined strains.
Small colony variant (SCV) is a phenotypic variant of
P. aeruginosa colonies, which are circular opaque dwarf colonies with a diameter about three times smaller than wild-type colonies, frequently obtained from samples of
P. aeruginosa-infected cystic fibrosis lung [
71]. The phage Pf4 is known to cause small colony variant (SCV) at the late stage of PAO1 biofilm development [
11,
13]. Moreover, increased
xisF4 gene expression in PAO1 can lead to higher Pf4 phage production, and then the number of SCV colonies increased by 55% [
72]. However, the percentage obtained in this study for SCV in lysogenic strains was below 10%, with no significant difference. Interestingly, PA14 strain carries Pf5 prophage, and its relatedness to SCV formation had not been proven previously [
12]. Based on the previous finding that SCVs lack twitching motility [
53], and the results that Pf4 decreases twitching motility, we expected to detect a higher percentage of SCV, which was not the case. It was obvious that Pf4 did not influence SCV production in PA14 and LESB58.
The infection with Pf4 filamentous phages can contribute to the increased antibiotic susceptibility of bacteria, and the phenomenon seems to be both strain- and antibiotic-dependent. Namely, the increase in antibiotic activity was obvious for ciprofloxacin, gentamicin, ceftazidime, and streptomycin, against Pf4-infected LESB58, and for ciprofloxacin and ceftazidime against Pf4-infected PA14. It is interesting to notice that sensitivity of both LESB58 and PA14 to ceftazidime changed from resistant to intermediately sensitive after Pf4 infection, indicating significant re-sensitization to this antimicrobial agent. Moreover, LESB58 changed sensitivity to streptomycin upon Pf4 infection from intermediately sensitive to sensitive, while in PA14, this change was from resistant to intermediate resistant. Finally, original LESB58 was intermediately sensitive to tetracycline, and became sensitive upon Pf4 infection.
As Pf4 contributes to phage enhanced growth in a form of biofilm and knowing that bacteria in biofilm are up to 1000 times resistant [
73], it can be expected that sensitivity will decrease, but results indicated the opposite. However, the method comprises immediate treatment of planktonic bacteria with antibiotics, so the antibiotic exhibits its activity prior to possibility of biofilm formation. The increased sensitivity upon filamentous phage infection was previously documented for other phage-host systems; e.g., Wang et al., (2013) [
74] found that Vibrio phage VEJ increased
Vibrio cholerae sensitivity to ampicillin. Hagens et al., (2006) [
75] indicated that filamentous phages in combination with small doses of antibiotics can inhibit and even kill
P. aeruginosa strains. Moreover, two different PAO1 strains, one harboring a gentamicin resistance plasmid and second to tetracycline, became more susceptible after the use of filamentous phages in combination with gentamicin and tetracycline. The authors hypothesized that pores for extrusion influence the host cell membrane permeability, so the membrane is less effective as a barrier against antibiotic penetration. Although this explanation seems reasonable, the pores for extrusion contain a multimeric tightly-gated channel through the membrane that does not allow molecule inflow into the cell [
76]. The cryo-electron microscopy revealed that the multimeric channel has a central pore 6.0–8.8 nm in diameter, but in the middle of the channel, the pore is tightly closed [
77]. The finding that susceptibility is increased for various antibiotics, however, supports the assumption that Pf4 phage changes cell permeability upon infection. Thus, another possible mechanism is that
Pseudomonas filamentous phages can change the membrane potential of cells, previously documented for Escherichia phage f1 [
78]. This change can consequently alter membrane permeability by affecting the function of certain channels in the outer membrane, as a result of phage protein integration into membranes prior to extrusion. Moreover, it can lead to the opening of channels for phage extrusion, allowing antibiotic inflow into cells. If the extrusion pores can be opened prior to phage extrusion, and if they can allow molecule inflow into a cell, should be revealed in future studies. Our findings open up a new possible approach in phage therapy—treatment of multidrug and pan drug resistant strains with filamentous phages, in order to re-sensitize bacteria to certain antibiotics. However, this approach has to be considered with caution in order to avoid unwanted lysogenic conversion and increased virulence of strains. The choice of phage should be focused on those that do not contribute to the formation of biofilm and that significantly increase the susceptibility of strains to antibiotics.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}