
Characterization of SARS-CoV-2 Evasion: Interferon Pathway and Therapeutic Options


Abstract
:1. Introduction
2. Evasion of Innate Immunity by SARS-CoV-2
2.1. Evasion of Sensing by Host Innate Immune Receptors
2.2. Inhibition of Innate Immune Receptor Signaling and IFN Production
2.3. Inhibition of IFN Signaling and ISG Expression
2.4. Inhibition of Host Protein Production by Targeting Post-Transcriptional and Translational Steps
3. Therapeutic Options
3.1. IFN-α
3.2. IFN-β
3.3. IFN-λ
4. Conclusions and Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CARD | caspase activation and recruitment domain |
| CCL2 | chemokine ligand 2 |
| 3CLpro | protease 3C-like |
| DMV | double-membrane vesicle |
| dsRNA | double-stranded RNA |
| E | envelope |
| FDA | United States Food and Drug Administration |
| IFN | interferon |
| IFNAR | IFN-α receptor |
| IL | interleukin |
| IFN-LR1 | IFN-l receptor 1 |
| IKKα/ß | inhibitor of nuclear factor kappa-B kinase |
| IRF | IFN-regulatory factor |
| ISG | IFN-stimulated gene |
| ISRE | IFN-stimulated response element |
| ISGF | IFN-stimulated gene factor |
| JAK | Janus-associated kinase |
| KPNA2 | importin karyopherin α 2 |
| M | membrane |
| MAVS | mitochondrial-associated viral signaling protein |
| MDA5 | melanoma differentiation-associated protein 5 |
| N | nucleocapsid |
| NEMO | NF-κB essential modulator |
| NF-kB | nuclear factor kB |
| NPC | nuclear pore complex |
| NSP | nonstructural protein |
| ORF | open reading frame |
| PAMP | pathogen-associated molecular patterns |
| PLpro | papain-like protease |
| PRR | pattern-recognition receptors |
| RIG-I | retinoic acid-inducible gene I protein |
| RdRp | RNA-dependent RNA polymerase |
| RT-qPCR | reverse transcription-quantitative PCR |
| SARS-CoV-2 | severe acute respiratory syndrome coronavirus 2 |
| sgRNA | subgenomic RNA |
| S | spike |
| SREBP1 | sterol regulatory element-binding protein 1 |
| SRP | signal recognition particle |
| STAT | transducer and activator of transcription |
| TBK1 | TANK-binding kinase 1 |
| TLR | Toll-like receptor |
| TNF | tumor necrosis factor |
| TRAF | TNF receptor-associated factor |
| TYK | tyrosine kinase |
| WHO | World Health Organization |
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khailany, R.A.; Safdar, M.; Ozaslan, M. Genomic Characterization of a Novel SARS-CoV-2. Gene Rep. 2020, 19, 100682. [Google Scholar] [CrossRef] [PubMed]
- Malone, B.; Urakova, N.; Snijder, E.J.; Campbell, E.A. Structures and Functions of Coronavirus Replication–Transcription Complexes and Their Relevance for SARS-CoV-2 Drug Design. Nat. Rev. Mol. Cell Biol. 2022, 23, 21–39. [Google Scholar] [CrossRef] [PubMed]
- Davidson, A.D.; Williamson, M.K.; Lewis, S.; Shoemark, D.; Carroll, M.W.; Heesom, K.J.; Zambon, M.; Ellis, J.; Lewis, P.A.; Hiscox, J.A.; et al. Characterisation of the Transcriptome and Proteome of SARS-CoV-2 Reveals a Cell Passage Induced in-Frame Deletion of the Furin-like Cleavage Site from the Spike Glycoprotein. Genome Med. 2020, 12, 68. [Google Scholar] [CrossRef]
- Kim, D.; Lee, J.-Y.; Yang, J.-S.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921.e10. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A Pneumonia Outbreak Associated with a New Coronavirus of Probable Bat Origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Hartenian, E.; Nandakumar, D.; Lari, A.; Ly, M.; Tucker, J.M.; Glaunsinger, B.A. The Molecular Virology of Coronaviruses. J. Biol. Chem. 2020, 295, 12910–12934. [Google Scholar] [CrossRef]
- Seth, R.B.; Sun, L.; Chen, Z.J. Antiviral Innate Immunity Pathways. Cell Res. 2006, 16, 141–147. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical Features of Patients Infected with 2019 Novel Coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Meng, Y.; Wang, K.; Zhang, X.; Chen, W.; Sheng, J.; Qiu, Y.; Diao, H.; Li, L. Inflammation and Antiviral Immune Response Associated with Severe Progression of COVID-19. Front. Immunol. 2021, 12, 631226. [Google Scholar] [CrossRef]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical Course and Risk Factors for Mortality of Adult Inpatients with COVID-19 in Wuhan, China: A Retrospective Cohort Study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Péré, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired Type I Interferon Activity and Inflammatory Responses in Severe COVID-19 Patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.-C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045. [Google Scholar] [CrossRef] [PubMed]
- Galani, I.-E.; Rovina, N.; Lampropoulou, V.; Triantafyllia, V.; Manioudaki, M.; Pavlos, E.; Koukaki, E.; Fragkou, P.C.; Panou, V.; Rapti, V.; et al. Untuned Antiviral Immunity in COVID-19 Revealed by Temporal Type I/III Interferon Patterns and Flu Comparison. Nat. Immunol. 2021, 22, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Brubaker, S.W.; Bonham, K.S.; Zanoni, I.; Kagan, J.C. Innate Immune Pattern Recognition: A Cell Biological Perspective. Annu. Rev. Immunol. 2015, 33, 257–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goubau, D.; Deddouche, S.; Reis e Sousa, C. Cytosolic Sensing of Viruses. Immunity 2013, 38, 855–869. [Google Scholar] [CrossRef] [Green Version]
- Carty, M.; Guy, C.; Bowie, A.G. Detection of Viral Infections by Innate Immunity. Biochem. Pharmacol. 2021, 183, 114316. [Google Scholar] [CrossRef]
- Reikine, S.; Nguyen, J.B.; Modis, Y. Pattern Recognition and Signaling Mechanisms of RIG-I and MDA5. Front. Immunol. 2014, 5, 342. [Google Scholar] [CrossRef] [Green Version]
- Kouwaki, T.; Nishimura, T.; Wang, G.; Oshiumi, H. RIG-I-Like Receptor-Mediated Recognition of Viral Genomic RNA of Severe Acute Respiratory Syndrome Coronavirus-2 and Viral Escape from the Host Innate Immune Responses. Front. Immunol. 2021, 12, 700926. [Google Scholar] [CrossRef]
- Thorne, L.G.; Reuschl, A.; Zuliani-Alvarez, L.; Whelan, M.V.X.; Turner, J.; Noursadeghi, M.; Jolly, C.; Towers, G.J. SARS-CoV-2 Sensing by RIG-I and MDA5 Links Epithelial Infection to Macrophage Inflammation. EMBO J. 2021, 40, e107826. [Google Scholar] [CrossRef]
- Yin, X.; Riva, L.; Pu, Y.; Martin-Sancho, L.; Kanamune, J.; Yamamoto, Y.; Sakai, K.; Gotoh, S.; Miorin, L.; De Jesus, P.D.; et al. MDA5 Governs the Innate Immune Response to SARS-CoV-2 in Lung Epithelial Cells. Cell Rep. 2021, 34, 108628. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, N.G.; Chauveau, L.; Hertzog, J.; Bridgeman, A.; Fowler, G.; Moonen, J.P.; Dupont, M.; Russell, R.A.; Noerenberg, M.; Rehwinkel, J. The RNA Sensor MDA5 Detects SARS-CoV-2 Infection. Sci. Rep. 2021, 11, 13638. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, A.; Yamada, T. Regulation of Signaling Mediated by Nucleic Acid Sensors for Innate Interferon-Mediated Responses during Viral Infection. Int. Immunol. 2019, 31, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Anaeigoudari, A.; Mollaei, H.R.; Arababadi, M.K.; Nosratabadi, R. Severe Acute Respiratory Syndrome Coronavirus 2: The Role of the Main Components of the Innate Immune System. Inflammation 2021, 44, 2151–2169. [Google Scholar] [CrossRef]
- Lazear, H.M.; Schoggins, J.W.; Diamond, M.S. Shared and Distinct Functions of Type I and Type III Interferons. Immunity 2019, 50, 907–923. [Google Scholar] [CrossRef]
- Schindler, C.; Levy, D.E.; Decker, T. JAK-STAT Signaling: From Interferons to Cytokines. J. Biol. Chem. 2007, 282, 20059–20063. [Google Scholar] [CrossRef] [Green Version]
- Mesev, E.V.; LeDesma, R.A.; Ploss, A. Decoding Type I and III Interferon Signalling during Viral Infection. Nat. Microbiol. 2019, 4, 914–924. [Google Scholar] [CrossRef]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-Stimulated Genes: A Complex Web of Host Defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [Green Version]
- Schoggins, J.W. Interferon-Stimulated Genes: What Do They All Do? Annu. Rev. Virol. 2019, 6, 567–584. [Google Scholar] [CrossRef]
- Hagemeijer, M.C.; Monastyrska, I.; Griffith, J.; van der Sluijs, P.; Voortman, J.; van Bergen en Henegouwen, P.M.; Vonk, A.M.; Rottier, P.J.M.; Reggiori, F.; de Haan, C.A.M. Membrane Rearrangements Mediated by Coronavirus Nonstructural Proteins 3 and 4. Virology 2014, 458, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Klein, S.; Cortese, M.; Winter, S.L.; Wachsmuth-Melm, M.; Neufeldt, C.J.; Cerikan, B.; Stanifer, M.L.; Boulant, S.; Bartenschlager, R.; Chlanda, P. SARS-CoV-2 Structure and Replication Characterized by in Situ Cryo-Electron Tomography. Nat. Commun. 2020, 11, 5885. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; Ruggiero, A.; Squeglia, F.; Maga, G.; Berisio, R. A Structural View of SARS-CoV-2 RNA Replication Machinery: RNA Synthesis, Proofreading and Final Capping. Cells 2020, 9, 1267. [Google Scholar] [CrossRef] [PubMed]
- Züst, R.; Cervantes-Barragan, L.; Habjan, M.; Maier, R.; Neuman, B.W.; Ziebuhr, J.; Szretter, K.J.; Baker, S.C.; Barchet, W.; Diamond, M.S.; et al. Ribose 2′-O-Methylation Provides a Molecular Signature for the Distinction of Self and Non-Self MRNA Dependent on the RNA Sensor Mda5. Nat. Immunol. 2011, 12, 137–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, D.W.; Amarasinghe, G.K. When Your Cap Matters: Structural Insights into Self vs Non-Self Recognition of 5′ RNA by Immunomodulatory Host Proteins. Curr. Opin. Struct. Biol. 2016, 36, 133–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Xiao, F.; Hu, D.; Ge, W.; Tian, M.; Wang, W.; Pan, P.; Wu, K.; Wu, J. SARS-CoV-2 Nucleocapsid Protein Interacts with RIG-I and Represses RIG-Mediated IFN-β Production. Viruses 2020, 13, 47. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like Protease Regulates SARS-CoV-2 Viral Spread and Innate Immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef]
- Liu, G.; Lee, J.-H.; Parker, Z.M.; Acharya, D.; Chiang, J.J.; van Gent, M.; Riedl, W.; Davis-Gardner, M.E.; Wies, E.; Chiang, C.; et al. ISG15-Dependent Activation of the Sensor MDA5 Is Antagonized by the SARS-CoV-2 Papain-like Protease to Evade Host Innate Immunity. Nat. Microbiol. 2021, 6, 467–478. [Google Scholar] [CrossRef]
- Liu, Y.; Qin, C.; Rao, Y.; Ngo, C.; Feng, J.J.; Zhao, J.; Zhang, S.; Wang, T.-Y.; Carriere, J.; Savas, A.C.; et al. SARS-CoV-2 Nsp5 Demonstrates Two Distinct Mechanisms Targeting RIG-I and MAVS To Evade the Innate Immune Response. mBio 2021, 12, e0233521. [Google Scholar] [CrossRef]
- Gori Savellini, G.; Anichini, G.; Gandolfo, C.; Cusi, M.G. SARS-CoV-2 N Protein Targets TRIM25-Mediated RIG-I Activation to Suppress Innate Immunity. Viruses 2021, 13, 1439. [Google Scholar] [CrossRef]
- Oh, S.J.; Shin, O.S. SARS-CoV-2 Nucleocapsid Protein Targets RIG-I-Like Receptor Pathways to Inhibit the Induction of Interferon Response. Cells 2021, 10, 530. [Google Scholar] [CrossRef]
- Zhao, Y.; Sui, L.; Wu, P.; Wang, W.; Wang, Z.; Yu, Y.; Hou, Z.; Tan, G.; Liu, Q.; Wang, G. A Dual-Role of SARS-CoV-2 Nucleocapsid Protein in Regulating Innate Immune Response. Signal Transduct. Target. Ther. 2021, 6, 331. [Google Scholar] [CrossRef] [PubMed]
- Yuen, C.-K.; Lam, J.-Y.; Wong, W.-M.; Mak, L.-F.; Wang, X.; Chu, H.; Cai, J.-P.; Jin, D.-Y.; To, K.K.-W.; Chan, J.F.-W.; et al. SARS-CoV-2 Nsp13, Nsp14, Nsp15 and Orf6 Function as Potent Interferon Antagonists. Emerg. Microbes Infect. 2020, 9, 1418–1428. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Dong, X.; Ma, R.; Wang, W.; Xiao, X.; Tian, Z.; Wang, C.; Wang, Y.; Li, L.; Ren, L.; et al. Activation and Evasion of Type I Interferon Responses by SARS-CoV-2. Nat. Commun. 2020, 11, 3810. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Cao, Z.; Xie, X.; Zhang, X.; Chen, J.Y.-C.; Wang, H.; Menachery, V.D.; Rajsbaum, R.; Shi, P.-Y. Evasion of Type I Interferon by SARS-CoV-2. Cell Rep. 2020, 33, 108234. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-Y.; Liao, C.-H.; Wang, Q.; Tan, Y.-J.; Luo, R.; Qiu, Y.; Ge, X.-Y. The ORF6, ORF8 and Nucleocapsid Proteins of SARS-CoV-2 Inhibit Type I Interferon Signaling Pathway. Virus Res. 2020, 286, 198074. [Google Scholar] [CrossRef]
- Shemesh, M.; Aktepe, T.E.; Deerain, J.M.; McAuley, J.L.; Audsley, M.D.; David, C.T.; Purcell, D.F.J.; Urin, V.; Hartmann, R.; Moseley, G.W.; et al. SARS-CoV-2 Suppresses IFNβ Production Mediated by NSP1, 5, 6, 15, ORF6 and ORF7b but Does Not Suppress the Effects of Added Interferon. PLoS Pathog. 2021, 17, e1009800. [Google Scholar] [CrossRef]
- Sato, M.; Suemori, H.; Hata, N.; Asagiri, M.; Ogasawara, K.; Nakao, K.; Nakaya, T.; Katsuki, M.; Noguchi, S.; Tanaka, N.; et al. Distinct and Essential Roles of Transcription Factors IRF-3 and IRF-7 in Response to Viruses for IFNa/b Gene Induction. Immunity 2000, 13, 538–548. [Google Scholar] [CrossRef] [Green Version]
- Sato, M.; Hata, N.; Asagiri, M.; Nakaya, T.; Taniguchi, T.; Tanaka, N. Positive Feedback Regulation of Type I IFN Genes by the IFN-Inducible Transcription Factor IRF-7. FEBS Lett. 1998, 441, 106–110. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Ishida, R.; Strilets, T.; Cole, J.; Lopez-Orozco, J.; Fayad, N.; Felix-Lopez, A.; Elaish, M.; Evseev, D.; Magor, K.E.; et al. SARS-CoV-2 Nonstructural Protein 1 Inhibits the Interferon Response by Causing Depletion of Key Host Signaling Factors. J. Virol. 2021, 95, e00266-21. [Google Scholar] [CrossRef]
- Wang, W.; Zhou, Z.; Xiao, X.; Tian, Z.; Dong, X.; Wang, C.; Li, L.; Ren, L.; Lei, X.; Xiang, Z.; et al. SARS-CoV-2 Nsp12 Attenuates Type I Interferon Production by Inhibiting IRF3 Nuclear Translocation. Cell. Mol. Immunol. 2021, 18, 945–953. [Google Scholar] [CrossRef]
- Konno, Y.; Kimura, I.; Uriu, K.; Fukushi, M.; Irie, T.; Koyanagi, Y.; Sauter, D.; Gifford, R.J.; Nakagawa, S.; Sato, K. SARS-CoV-2 ORF3b Is a Potent Interferon Antagonist Whose Activity Is Increased by a Naturally Occurring Elongation Variant. Cell Rep. 2020, 32, 108185. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Zhao, K.; Zhang, B.; Hua, R.; Fang, Y.; Jiang, W.; Zhang, J.; Hui, L.; Zheng, Y.; Li, Y.; et al. SARS-CoV-2 NSP12 Protein Is Not an Interferon-β Antagonist. J. Virol. 2021, 95, e00747-21. [Google Scholar] [CrossRef] [PubMed]
- Loo, Y.-M.; Gale, M. Immune Signaling by RIG-I-like Receptors. Immunity 2011, 34, 680–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Y.-Z.; Wang, S.-Y.; Zheng, Z.-Q.; Huang, Y.; Li, W.-W.; Xu, Z.-S.; Wang, Y.-Y. SARS-CoV-2 Membrane Glycoprotein M Antagonizes the MAVS-Mediated Innate Antiviral Response. Cell. Mol. Immunol. 2021, 18, 613–620. [Google Scholar] [CrossRef]
- Sui, L.; Zhao, Y.; Wang, W.; Wu, P.; Wang, Z.; Yu, Y.; Hou, Z.; Tan, G.; Liu, Q. SARS-CoV-2 Membrane Protein Inhibits Type I Interferon Production Through Ubiquitin-Mediated Degradation of TBK1. Front. Immunol. 2021, 12, 662989. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhuang, M.-W.; Han, L.; Zhang, J.; Nan, M.-L.; Zhan, P.; Kang, D.; Liu, X.; Gao, C.; Wang, P.-H. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Membrane (M) Protein Inhibits Type I and III Interferon Production by Targeting RIG-I/MDA-5 Signaling. Signal Transduct. Target. Ther. 2020, 5, 299. [Google Scholar] [CrossRef]
- Vazquez, C.; Swanson, S.E.; Negatu, S.G.; Dittmar, M.; Miller, J.; Ramage, H.R.; Cherry, S.; Jurado, K.A. SARS-CoV-2 Viral Proteins NSP1 and NSP13 Inhibit Interferon Activation through Distinct Mechanisms. PLoS ONE 2021, 16, e0253089. [Google Scholar] [CrossRef]
- Han, L.; Zhuang, M.-W.; Deng, J.; Zheng, Y.; Zhang, J.; Nan, M.-L.; Zhang, X.-J.; Gao, C.; Wang, P.-H. SARS-CoV-2 ORF9b Antagonizes Type I and III Interferons by Targeting Multiple Components of the RIG-I/MDA-5-MAVS, TLR3-TRIF, and CGAS-STING Signaling Pathways. J. Med. Virol. 2021, 93, 5376–5389. [Google Scholar] [CrossRef]
- Jiang, H.-W.; Zhang, H.-N.; Meng, Q.-F.; Xie, J.; Li, Y.; Chen, H.; Zheng, Y.-X.; Wang, X.-N.; Qi, H.; Zhang, J.; et al. SARS-CoV-2 Orf9b Suppresses Type I Interferon Responses by Targeting TOM70. Cell. Mol. Immunol. 2020, 17, 998–1000. [Google Scholar] [CrossRef]
- Wu, J.; Shi, Y.; Pan, X.; Wu, S.; Hou, R.; Zhang, Y.; Zhong, T.; Tang, H.; Du, W.; Wang, L.; et al. SARS-CoV-2 ORF9b Inhibits RIG-I-MAVS Antiviral Signaling by Interrupting K63-Linked Ubiquitination of NEMO. Cell Rep. 2021, 34, 108761. [Google Scholar] [CrossRef]
- Makiyama, K.; Hazawa, M.; Kobayashi, A.; Lim, K.; Voon, D.C.; Wong, R.W. NSP9 of SARS-CoV-2 Attenuates Nuclear Transport by Hampering Nucleoporin 62 Dynamics and Functions in Host Cells. Biochem. Biophys. Res. Commun. 2022, 586, 137–142. [Google Scholar] [CrossRef]
- Moustaqil, M.; Ollivier, E.; Chiu, H.-P.; Van Tol, S.; Rudolffi-Soto, P.; Stevens, C.; Bhumkar, A.; Hunter, D.J.B.; Freiberg, A.N.; Jacques, D.; et al. SARS-CoV-2 Proteases PLpro and 3CLpro Cleave IRF3 and Critical Modulators of Inflammatory Pathways (NLRP12 and TAB1): Implications for Disease Presentation across Species. Emerg. Microbes Infect. 2021, 10, 178–195. [Google Scholar] [CrossRef] [PubMed]
- Fung, S.-Y.; Siu, K.-L.; Lin, H.; Yeung, M.L.; Jin, D.-Y. SARS-CoV-2 Main Protease Suppresses Type I Interferon Production by Preventing Nuclear Translocation of Phosphorylated IRF3. Int. J. Biol. Sci. 2021, 17, 1547–1554. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ma, Z.; Wu, Y.; Shi, X.; Zhang, Y.; Zhang, M.; Zhang, M.; Wang, L.; Liu, W. SARS-CoV-2 3C-like Protease Antagonizes Interferon-Beta Production by Facilitating the Degradation of IRF3. Cytokine 2021, 148, 155697. [Google Scholar] [CrossRef] [PubMed]
- Hayn, M.; Hirschenberger, M.; Koepke, L.; Nchioua, R.; Straub, J.H.; Klute, S.; Hunszinger, V.; Zech, F.; Prelli Bozzo, C.; Aftab, W.; et al. Systematic Functional Analysis of SARS-CoV-2 Proteins Uncovers Viral Innate Immune Antagonists and Remaining Vulnerabilities. Cell Rep. 2021, 35, 109126. [Google Scholar] [CrossRef]
- Kimura, I.; Konno, Y.; Uriu, K.; Hopfensperger, K.; Sauter, D.; Nakagawa, S.; Sato, K. Sarbecovirus ORF6 Proteins Hamper Induction of Interferon Signaling. Cell Rep. 2021, 34, 108916. [Google Scholar] [CrossRef]
- Miorin, L.; Kehrer, T.; Sanchez-Aparicio, M.T.; Zhang, K.; Cohen, P.; Patel, R.S.; Cupic, A.; Makio, T.; Mei, M.; Moreno, E.; et al. SARS-CoV-2 Orf6 Hijacks Nup98 to Block STAT Nuclear Import and Antagonize Interferon Signaling. Proc. Natl. Acad. Sci. USA 2020, 117, 28344–28354. [Google Scholar] [CrossRef]
- Addetia, A.; Lieberman, N.A.P.; Phung, Q.; Hsiang, T.-Y.; Xie, H.; Roychoudhury, P.; Shrestha, L.; Loprieno, M.A.; Huang, M.-L.; Gale, M.; et al. SARS-CoV-2 ORF6 Disrupts Bidirectional Nucleocytoplasmic Transport through Interactions with Rae1 and Nup98. mBio 2021, 12, e00065-21. [Google Scholar] [CrossRef]
- Mu, J.; Fang, Y.; Yang, Q.; Shu, T.; Wang, A.; Huang, M.; Jin, L.; Deng, F.; Qiu, Y.; Zhou, X. SARS-CoV-2 N Protein Antagonizes Type I Interferon Signaling by Suppressing Phosphorylation and Nuclear Translocation of STAT1 and STAT2. Cell Discov. 2020, 6, 65. [Google Scholar] [CrossRef]
- Cao, Z.; Xia, H.; Rajsbaum, R.; Xia, X.; Wang, H.; Shi, P.-Y. Ubiquitination of SARS-CoV-2 ORF7a Promotes Antagonism of Interferon Response. Cell. Mol. Immunol. 2021, 18, 746–748. [Google Scholar] [CrossRef]
- Banerjee, A.K.; Blanco, M.R.; Bruce, E.A.; Honson, D.D.; Chen, L.M.; Chow, A.; Bhat, P.; Ollikainen, N.; Quinodoz, S.A.; Loney, C.; et al. SARS-CoV-2 Disrupts Splicing, Translation, and Protein Trafficking to Suppress Host Defenses. Cell 2020, 183, 1325–1339. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.M.; Clair, L.A.S.; Perera, R.; Parker, R. SARS-CoV-2 Infection Triggers Widespread Host MRNA Decay Leading to an MRNA Export Block. RNA 2021, 27, 1318–1329. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Miorin, L.; Makio, T.; Dehghan, I.; Gao, S.; Xie, Y.; Zhong, H.; Esparza, M.; Kehrer, T.; Kumar, A.; et al. Nsp1 Protein of SARS-CoV-2 Disrupts the MRNA Export Machinery to Inhibit Host Gene Expression. Sci. Adv. 2021, 7, eabe7386. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Peng, L.; Park, J.J.; Hu, Y.; Devarkar, S.C.; Dong, M.B.; Shen, Q.; Wu, S.; Chen, S.; Lomakin, I.B.; et al. Nonstructural Protein 1 of SARS-CoV-2 Is a Potent Pathogenicity Factor Redirecting Host Protein Synthesis Machinery toward Viral RNA. Mol. Cell 2020, 80, 1055–1066. [Google Scholar] [CrossRef] [PubMed]
- Thoms, M.; Buschauer, R.; Ameismeier, M.; Koepke, L.; Denk, T.; Hirschenberger, M.; Kratzat, H.; Hayn, M.; Mackens-Kiani, T.; Cheng, J.; et al. Structural Basis for Translational Shutdown and Immune Evasion by the Nsp1 Protein of SARS-CoV-2. Science 2020, 369, 1249–1255. [Google Scholar] [CrossRef] [PubMed]
- Schubert, K.; Karousis, E.D.; Jomaa, A.; Scaiola, A.; Echeverria, B.; Gurzeler, L.-A.; Leibundgut, M.; Thiel, V.; Mühlemann, O.; Ban, N. SARS-CoV-2 Nsp1 Binds the Ribosomal MRNA Channel to Inhibit Translation. Nat. Struct. Mol. Biol. 2020, 27, 959–966. [Google Scholar] [CrossRef]
- Lapointe, C.P.; Grosely, R.; Johnson, A.G.; Wang, J.; Fernández, I.S.; Puglisi, J.D. Dynamic Competition between SARS-CoV-2 NSP1 and MRNA on the Human Ribosome Inhibits Translation Initiation. Proc. Natl. Acad. Sci. USA 2021, 118, e2017715118. [Google Scholar] [CrossRef]
- Mendez, A.S.; Ly, M.; González-Sánchez, A.M.; Hartenian, E.; Ingolia, N.T.; Cate, J.H.; Glaunsinger, B.A. The N-Terminal Domain of SARS-CoV-2 Nsp1 Plays Key Roles in Suppression of Cellular Gene Expression and Preservation of Viral Gene Expression. Cell Rep. 2021, 37, 109841. [Google Scholar] [CrossRef]
- Trouillet-Assant, S.; Viel, S.; Gaymard, A.; Pons, S.; Richard, J.-C.; Perret, M.; Villard, M.; Brengel-Pesce, K.; Lina, B.; Mezidi, M.; et al. Type I IFN Immunoprofiling in COVID-19 Patients. J. Allergy Clin. Immunol. 2020, 146, 206–208. [Google Scholar] [CrossRef]
- Contoli, M.; Papi, A.; Tomassetti, L.; Rizzo, P.; Vieceli Dalla Sega, F.; Fortini, F.; Torsani, F.; Morandi, L.; Ronzoni, L.; Zucchetti, O.; et al. Blood Interferon-α Levels and Severity, Outcomes, and Inflammatory Profiles in Hospitalized COVID-19 Patients. Front. Immunol. 2021, 12, 648004. [Google Scholar] [CrossRef]
- Zhang, Q.; Bastard, P.; Liu, Z.; Le Pen, J.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Sabli, I.K.D.; Hodeib, S.; Korol, C.; et al. Inborn Errors of Type I IFN Immunity in Patients with Life-Threatening COVID-19. Science 2020, 370, eabd4570. [Google Scholar] [CrossRef] [PubMed]
- Van der Made, C.I.; Simons, A.; Schuurs-Hoeijmakers, J.; van den Heuvel, G.; Mantere, T.; Kersten, S.; van Deuren, R.C.; Steehouwer, M.; van Reijmersdal, S.V.; Jaeger, M.; et al. Presence of Genetic Variants Among Young Men with Severe COVID-19. JAMA 2020, 324, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Abolhassani, H.; Vosughimotlagh, A.; Asano, T.; Landegren, N.; Boisson, B.; Delavari, S.; Bastard, P.; Aranda-Guillén, M.; Wang, Y.; Zuo, F.; et al. X-Linked TLR7 Deficiency Underlies Critical COVID-19 Pneumonia in a Male Patient with Ataxia-Telangiectasia. J. Clin. Immunol. 2022, 42, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.-H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Béziat, V.; et al. Autoantibodies against Type I IFNs in Patients with Life-Threatening COVID-19. Science 2020, 370, eabd4585. [Google Scholar] [CrossRef]
- Koning, R.; Bastard, P.; Casanova, J.-L.; Brouwer, M.C.; van de Beek, D. Autoantibodies against Type I Interferons Are Associated with Multi-Organ Failure in COVID-19 Patients. Intensive Care Med. 2021, 47, 704–706. [Google Scholar] [CrossRef]
- Troya, J.; Bastard, P.; Planas-Serra, L.; Ryan, P.; Ruiz, M.; de Carranza, M.; Torres, J.; Martínez, A.; Abel, L.; Casanova, J.-L.; et al. Neutralizing Autoantibodies to Type I IFNs in >10% of Patients with Severe COVID-19 Pneumonia Hospitalized in Madrid, Spain. J. Clin. Immunol. 2021, 41, 914–922. [Google Scholar] [CrossRef]
- Van der Wijst, M.G.P.; Vazquez, S.E.; Hartoularos, G.C.; Bastard, P.; Grant, T.; Bueno, R.; Lee, D.S.; Greenland, J.R.; Sun, Y.; Perez, R.; et al. Type I Interferon Autoantibodies Are Associated with Systemic Immune Alterations in Patients with COVID-19. Sci. Transl. Med. 2021, 13, eabh2624. [Google Scholar] [CrossRef]
- Bastard, P.; Gervais, A.; Le Voyer, T.; Rosain, J.; Philippot, Q.; Manry, J.; Michailidis, E.; Hoffmann, H.-H.; Eto, S.; Garcia-Prat, M.; et al. Autoantibodies Neutralizing Type I IFNs Are Present in ~4% of Uninfected Individuals over 70 Years and Account for ~20% of COVID-19 Deaths. Sci. Immunol. 2021, 6, eabl4340. [Google Scholar] [CrossRef]
- Felgenhauer, U.; Schoen, A.; Gad, H.H.; Hartmann, R.; Schaubmar, A.R.; Failing, K.; Drosten, C.; Weber, F. Inhibition of SARS-CoV-2 by Type I and Type III Interferons. J. Biol. Chem. 2020, 295, 13958–13964. [Google Scholar] [CrossRef]
- Mantlo, E.; Bukreyeva, N.; Maruyama, J.; Paessler, S.; Huang, C. Antiviral Activities of Type I Interferons to SARS-CoV-2 Infection. Antiviral Res. 2020, 179, 104811. [Google Scholar] [CrossRef]
- Dinnon, K.H.; Leist, S.R.; Schäfer, A.; Edwards, C.E.; Martinez, D.R.; Montgomery, S.A.; West, A.; Yount, B.L.; Hou, Y.J.; Adams, L.E.; et al. A Mouse-Adapted Model of SARS-CoV-2 to Test COVID-19 Countermeasures. Nature 2020, 586, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Vanderheiden, A.; Ralfs, P.; Chirkova, T.; Upadhyay, A.A.; Zimmerman, M.G.; Bedoya, S.; Aoued, H.; Tharp, G.M.; Pellegrini, K.L.; Manfredi, C.; et al. Type I and Type III Interferons Restrict SARS-CoV-2 Infection of Human Airway Epithelial Cultures. J. Virol. 2020, 94, e00985-20. [Google Scholar] [CrossRef] [PubMed]
- Hoagland, D.A.; Møller, R.; Uhl, S.A.; Oishi, K.; Frere, J.; Golynker, I.; Horiuchi, S.; Panis, M.; Blanco-Melo, D.; Sachs, D.; et al. Leveraging the Antiviral Type I Interferon System as a First Line of Defense against SARS-CoV-2 Pathogenicity. Immunity 2021, 54, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Aghemo, A.; Rumi, M.G.; Colombo, M. Pegylated Interferons A2a and A2b in the Treatment of Chronic Hepatitis C. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 485–494. [Google Scholar] [CrossRef]
- Broquetas, T.; Garcia-Retortillo, M.; Micó, M.; Canillas, L.; Puigvehí, M.; Cañete, N.; Coll, S.; Viu, A.; Hernandez, J.J.; Bessa, X.; et al. Hepatitis B Surface Antigen and Hepatitis B Core-Related Antigen Kinetics after Adding Pegylated-Interferon to Nucleos(t)Ids Analogues in Hepatitis B e Antigen-Negative Patients. World J. Hepatol. 2020, 12, 1076–1088. [Google Scholar] [CrossRef]
- Zhou, Q.; Chen, V.; Shannon, C.P.; Wei, X.-S.; Xiang, X.; Wang, X.; Wang, Z.-H.; Tebbutt, S.J.; Kollmann, T.R.; Fish, E.N. Interferon-A2b Treatment for COVID-19. Front. Immunol. 2020, 11, 1061. [Google Scholar] [CrossRef]
- Pandit, A.; Bhalani, N.; Bhushan, B.L.S.; Koradia, P.; Gargiya, S.; Bhomia, V.; Kansagra, K. Efficacy and Safety of Pegylated Interferon Alfa-2b in Moderate COVID-19: A Phase II, Randomized, Controlled, Open-Label Study. Int. J. Infect. Dis. 2021, 105, 516–521. [Google Scholar] [CrossRef]
- Bhushan, B.L.S.; Wanve, S.; Koradia, P.; Bhomia, V.; Soni, P.; Chakraborty, S.; Khobragade, A.; Joshi, S.; Mendiratta, S.K.; Kansagra, K.K.; et al. Efficacy and Safety of Pegylated Interferon-A2b in Moderate COVID-19: A Phase 3, Randomized, Comparator-Controlled, Open-Label Study. Int. J. Infect. Dis. 2021, 111, 281–287. [Google Scholar] [CrossRef]
- Wang, B.; Li, D.; Liu, T.; Wang, H.; Luo, F.; Liu, Y. Subcutaneous Injection of IFN Alpha-2b for COVID-19: An Observational Study. BMC Infect. Dis. 2020, 20, 723. [Google Scholar] [CrossRef]
- Wang, N.; Zhan, Y.; Zhu, L.; Hou, Z.; Liu, F.; Song, P.; Qiu, F.; Wang, X.; Zou, X.; Wan, D.; et al. Retrospective Multicenter Cohort Study Shows Early Interferon Therapy Is Associated with Favorable Clinical Responses in COVID-19 Patients. Cell Host Microbe 2020, 28, 455–464. [Google Scholar] [CrossRef]
- Yu, J.; Lu, X.; Tong, L.; Shi, X.; Ma, J.; Lv, F.; Wu, J.; Pan, Q.; Yang, J.; Cao, H.; et al. Interferon-α-2b Aerosol Inhalation Is Associated with Improved Clinical Outcomes in Patients with Coronavirus Disease-2019. Br. J. Clin. Pharmacol. 2021, 87, 4737–4746. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, C.G.K.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M.; et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell 2020, 181, 1016–1035. [Google Scholar] [CrossRef]
- Blume, C.; Jackson, C.L.; Spalluto, C.M.; Legebeke, J.; Nazlamova, L.; Conforti, F.; Perotin, J.-M.; Frank, M.; Butler, J.; Crispin, M.; et al. A Novel ACE2 Isoform Is Expressed in Human Respiratory Epithelia and Is Upregulated in Response to Interferons and RNA Respiratory Virus Infection. Nat. Genet. 2021, 53, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.W.; Attig, J.; Bolland, W.; Young, G.R.; Major, J.; Wrobel, A.G.; Gamblin, S.; Wack, A.; Kassiotis, G. Tissue-Specific and Interferon-Inducible Expression of Nonfunctional ACE2 through Endogenous Retroelement Co-Option. Nat. Genet. 2020, 52, 1294–1302. [Google Scholar] [CrossRef] [PubMed]
- Onabajo, O.O.; Banday, A.R.; Yan, W.; Obajemu, A.; Stanifer, M.L.; Santer, D.M.; Florez-Vargas, O.; Piontkivska, H.; Vargas, J.; Kee, C.; et al. Interferons and Viruses Induce a Novel Truncated ACE2 Isoform and Not the full-length SARS-CoV-2 Receptor. Nat. Genet. 2020, 52, 1283–1293. [Google Scholar] [CrossRef] [PubMed]
- Kiss, J.; Yegutkin, G.G.; Koskinen, K.; Savunen, T.; Jalkanen, S.; Salmi, M. IFN-β Protects from Vascular Leakage via up-Regulation of CD73. Eur. J. Immunol. 2007, 37, 3334–3338. [Google Scholar] [CrossRef]
- Aeffner, F.; Woods, P.S.; Davis, I.C. Activation of A1-Adenosine Receptors Promotes Leukocyte Recruitment to the Lung and Attenuates Acute Lung Injury in Mice Infected with Influenza A/WSN/33 (H1N1) Virus. J. Virol. 2014, 88, 10214–10227. [Google Scholar] [CrossRef] [Green Version]
- Ranieri, V.M.; Pettilä, V.; Karvonen, M.K.; Jalkanen, J.; Nightingale, P.; Brealey, D.; Mancebo, J.; Ferrer, R.; Mercat, A.; Patroniti, N.; et al. Effect of Intravenous Interferon β-1a on Death and Days Free from Mechanical Ventilation Among Patients with Moderate to Severe Acute Respiratory Distress Syndrome: A Randomized Clinical Trial. JAMA 2020, 323, 725–733. [Google Scholar] [CrossRef]
- Hung, I.F.-N.; Lung, K.-C.; Tso, E.Y.-K.; Liu, R.; Chung, T.W.-H.; Chu, M.-Y.; Ng, Y.-Y.; Lo, J.; Chan, J.; Tam, A.R.; et al. Triple Combination of Interferon Beta-1b, Lopinavir–Ritonavir, and Ribavirin in the Treatment of Patients Admitted to Hospital with COVID-19: An Open-Label, Randomised, Phase 2 Trial. Lancet 2020, 395, 1695–1704. [Google Scholar] [CrossRef]
- Alavi Darazam, I.; Shokouhi, S.; Pourhoseingholi, M.A.; Naghibi Irvani, S.S.; Mokhtari, M.; Shabani, M.; Amirdosara, M.; Torabinavid, P.; Golmohammadi, M.; Hashemi, S.; et al. Role of Interferon Therapy in Severe COVID-19: The COVIFERON Randomized Controlled Trial. Sci. Rep. 2021, 11, 8059. [Google Scholar] [CrossRef]
- Davoudi-Monfared, E.; Rahmani, H.; Khalili, H.; Hajiabdolbaghi, M.; Salehi, M.; Abbasian, L.; Kazemzadeh, H.; Yekaninejad, M.S. A Randomized Clinical Trial of the Efficacy and Safety of Interferon β-1a in Treatment of Severe COVID-19. Antimicrob. Agents Chemother. 2020, 64, e01061-20. [Google Scholar] [CrossRef] [PubMed]
- WHO Solidarity Trial Consortium. Repurposed Antiviral Drugs for COVID-19—Interim WHO Solidarity Trial Results. N. Engl. J. Med. 2021, 384, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Kalil, A.C.; Mehta, A.K.; Patterson, T.F.; Erdmann, N.; Gomez, C.A.; Jain, M.K.; Wolfe, C.R.; Ruiz-Palacios, G.M.; Kline, S.; Regalado Pineda, J.; et al. Efficacy of Interferon Beta-1a plus Remdesivir Compared with Remdesivir Alone in Hospitalised Adults with COVID-19: A Double-Bind, Randomised, Placebo-Controlled, Phase 3 Trial. Lancet Respir. Med. 2021, 9, 1365–1376. [Google Scholar] [CrossRef]
- National Institutes of Health. Coronavirus Disease 2019 (COVID-19) Treatment Guidelines; National Institutes of Health: Bethesda, MD, USA, 2022; p. 394. [Google Scholar]
- Monk, P.D.; Marsden, R.J.; Tear, V.J.; Brookes, J.; Batten, T.N.; Mankowski, M.; Gabbay, F.J.; Davies, D.E.; Holgate, S.T.; Ho, L.-P.; et al. Safety and Efficacy of Inhaled Nebulised Interferon Beta-1a (SNG001) for Treatment of SARS-CoV-2 Infection: A Randomised, Double-Blind, Placebo-Controlled, Phase 2 Trial. Lancet Respir. Med. 2021, 9, 196–206. [Google Scholar] [CrossRef]
- Khamis, F.; Al Naabi, H.; Al Lawati, A.; Ambusaidi, Z.; Al Sharji, M.; Al Barwani, U.; Pandak, N.; Al Balushi, Z.; Al Bahrani, M.; Al Salmi, I.; et al. Randomized Controlled Open Label Trial on the Use of Favipiravir Combined with Inhaled Interferon Beta-1b in Hospitalized Patients with Moderate to Severe COVID-19 Pneumonia. Int. J. Infect. Dis. 2021, 102, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Klinkhammer, J.; Schnepf, D.; Ye, L.; Schwaderlapp, M.; Gad, H.H.; Hartmann, R.; Garcin, D.; Mahlakõiv, T.; Staeheli, P. IFN-λ Prevents Influenza Virus Spread from the Upper Airways to the Lungs and Limits Virus Transmission. eLife 2018, 7, e33354. [Google Scholar] [CrossRef]
- Davidson, S.; McCabe, T.M.; Crotta, S.; Gad, H.H.; Hessel, E.M.; Beinke, S.; Hartmann, R.; Wack, A. IFNλ Is a Potent Anti-influenza Therapeutic without the Inflammatory Side Effects of IFNα Treatment. EMBO Mol. Med. 2016, 8, 1099–1112. [Google Scholar] [CrossRef]
- Forero, A.; Ozarkar, S.; Li, H.; Lee, C.H.; Hemann, E.A.; Nadjsombati, M.S.; Hendricks, M.R.; So, L.; Green, R.; Roy, C.N.; et al. Differential Activation of the Transcription Factor IRF1 Underlies the Distinct Immune Responses Elicited by Type I and Type III Interferons. Immunity 2019, 51, 451–464. [Google Scholar] [CrossRef]
- Galani, I.E.; Triantafyllia, V.; Eleminiadou, E.-E.; Koltsida, O.; Stavropoulos, A.; Manioudaki, M.; Thanos, D.; Doyle, S.E.; Kotenko, S.V.; Thanopoulou, K.; et al. Interferon-λ Mediates Non-Redundant Front-Line Antiviral Protection against Influenza Virus Infection without Compromising Host Fitness. Immunity 2017, 46, 875–890.e6. [Google Scholar] [CrossRef]
- Broggi, A.; Tan, Y.; Granucci, F.; Zanoni, I. IFN-λ Suppresses Intestinal Inflammation by Non-Translational Regulation of Neutrophil Function. Nat. Immunol. 2017, 18, 1084–1093. [Google Scholar] [CrossRef]
- Blazek, K.; Eames, H.L.; Weiss, M.; Byrne, A.J.; Perocheau, D.; Pease, J.E.; Doyle, S.; McCann, F.; Williams, R.O.; Udalova, I.A. IFN-λ Resolves Inflammation via Suppression of Neutrophil Infiltration and IL-1β Production. J. Exp. Med. 2015, 212, 845–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanifer, M.L.; Kee, C.; Cortese, M.; Zumaran, C.M.; Triana, S.; Mukenhirn, M.; Kraeusslich, H.-G.; Alexandrov, T.; Bartenschlager, R.; Boulant, S. Critical Role of Type III Interferon in Controlling SARS-CoV-2 Infection in Human Intestinal Epithelial Cells. Cell Rep. 2020, 32, 107863. [Google Scholar] [CrossRef] [PubMed]
- Sohn, S.-Y.; Hearing, J.; Mugavero, J.; Kirillov, V.; Gorbunova, E.; Helminiak, L.; Mishra, S.; Mackow, E.; Hearing, P.; Reich, N.C.; et al. Interferon-Lambda Intranasal Protection and Differential Sex Pathology in a Murine Model of SARS-CoV-2 Infection. mBio 2021, 12, e02756-21. [Google Scholar] [CrossRef] [PubMed]
- Liou, T.G.; Adler, F.R.; Cahill, B.C.; Cox, D.R.; Cox, J.E.; Grant, G.J.; Hanson, K.E.; Hartsell, S.C.; Hatton, N.D.; Helms, M.N.; et al. SARS-CoV-2 Innate Effector Associations and Viral Load in Early Nasopharyngeal Infection. Physiol. Rep. 2021, 9, e14761. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, Y.; Homma, T.; Inoue, H.; Onitsuka, C.; Ikeda, H.; Goto, Y.; Sato, Y.; Kimura, T.; Hirai, K.; Ohta, S.; et al. Downregulation of Type III Interferons in Patients with Severe COVID-19. J. Med. Virol. 2021, 93, 4559–4563. [Google Scholar] [CrossRef]
- Shahbazi, M.; Amri Maleh, P.; Bagherzadeh, M.; Moulana, Z.; Sepidarkish, M.; Rezanejad, M.; Mirzakhani, M.; Ebrahimpour, S.; Ghorbani, H.; Ahmadnia, Z.; et al. Linkage of Lambda Interferons in Protection Against Severe COVID-19. J. Interferon Cytokine Res. 2021, 41, 149–152. [Google Scholar] [CrossRef]
- Fukuda, Y.; Homma, T.; Inoue, H.; Goto, Y.; Sato, Y.; Ikeda, H.; Onitsuka, C.; Sato, H.; Akimoto, K.; Ebato, T.; et al. Serum IL-28A/IFN-Λ2 Is Linked to Disease Severity of COVID-19. Sci. Rep. 2022, 12, 5458. [Google Scholar] [CrossRef]
- O’Brien, T.R.; Thomas, D.L.; Jackson, S.S.; Prokunina-Olsson, L.; Donnelly, R.P.; Hartmann, R. Weak Induction of Interferon Expression by Severe Acute Respiratory Syndrome Coronavirus 2 Supports Clinical Trials of Interferon-λ to Treat Early Coronavirus Disease 2019. Clin. Infect. Dis. 2020, 71, 1410–1412. [Google Scholar] [CrossRef]
- Feld, J.J.; Kandel, C.; Biondi, M.J.; Kozak, R.A.; Zahoor, M.A.; Lemieux, C.; Borgia, S.M.; Boggild, A.K.; Powis, J.; McCready, J.; et al. Peginterferon Lambda for the Treatment of Outpatients with COVID-19: A Phase 2, Placebo-Controlled Randomised Trial. Lancet Respir. Med. 2021, 9, 498–510. [Google Scholar] [CrossRef]
- Jagannathan, P.; Andrews, J.R.; Bonilla, H.; Hedlin, H.; Jacobson, K.B.; Balasubramanian, V.; Purington, N.; Kamble, S.; de Vries, C.R.; Quintero, O.; et al. Peginterferon Lambda-1a for Treatment of Outpatients with Uncomplicated COVID-19: A Randomized Placebo-Controlled Trial. Nat. Commun. 2021, 12, 1967. [Google Scholar] [CrossRef]
- Planet, P.J.; Parker, D.; Cohen, T.S.; Smith, H.; Leon, J.D.; Ryan, C.; Hammer, T.J.; Fierer, N.; Chen, E.I.; Prince, A.S. Lambda Interferon Restructures the Nasal Microbiome and Increases Susceptibility to Staphylococcus Aureus Superinfection. mBio 2016, 7, e01939-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rich, H.E.; McCourt, C.C.; Zheng, W.Q.; McHugh, K.J.; Robinson, K.M.; Wang, J.; Alcorn, J.F. Interferon Lambda Inhibits Bacterial Uptake during Influenza Superinfection. Infect. Immun. 2019, 87, e00114-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Saccon, E.; Appelberg, K.S.; Mikaeloff, F.; Rodriguez, J.E.; Vinhas, B.S.; Frisan, T.; Végvári, Á.; Mirazimi, A.; Neogi, U.; et al. Type-I Interferon Signatures in SARS-CoV-2 Infected Huh7 Cells. Cell Death Discov. 2021, 7, 114. [Google Scholar] [CrossRef] [PubMed]
- Lokugamage, K.G.; Hage, A.; de Vries, M.; Valero-Jimenez, A.M.; Schindewolf, C.; Dittmann, M.; Rajsbaum, R.; Menachery, V.D. Type I Interferon Susceptibility Distinguishes SARS-CoV-2 from SARS-CoV. J. Virol. 2020, 94, e01410-20. [Google Scholar] [CrossRef]
- Bessière, P.; Wasniewski, M.; Picard-Meyer, E.; Servat, A.; Figueroa, T.; Foret-Lucas, C.; Coggon, A.; Lesellier, S.; Boué, F.; Cebron, N.; et al. Intranasal Type I Interferon Treatment Is Beneficial Only When Administered before Clinical Signs Onset in the SARS-CoV-2 Hamster Model. PLOS Pathog. 2021, 17, e1009427. [Google Scholar] [CrossRef]
- Sposito, B.; Broggi, A.; Pandolfi, L.; Crotta, S.; Clementi, N.; Ferrarese, R.; Sisti, S.; Criscuolo, E.; Spreafico, R.; Long, J.M.; et al. The Interferon Landscape along the Respiratory Tract Impacts the Severity of COVID-19. Cell 2021, 184, 4953–4968. [Google Scholar] [CrossRef]
- Ziegler, C.G.K.; Miao, V.N.; Owings, A.H.; Navia, A.W.; Tang, Y.; Bromley, J.D.; Lotfy, P.; Sloan, M.; Laird, H.; Williams, H.B.; et al. Impaired Local Intrinsic Immunity to SARS-CoV-2 Infection in Severe COVID-19. Cell 2021, 184, 4713–4733. [Google Scholar] [CrossRef]
- Lucas, C.; Wong, P.; Klein, J.; Castro, T.B.R.; Silva, J.; Sundaram, M.; Ellingson, M.K.; Mao, T.; Oh, J.E.; Israelow, B.; et al. Longitudinal Analyses Reveal Immunological Misfiring in Severe COVID-19. Nature 2020, 584, 463–469. [Google Scholar] [CrossRef]
- Lee, J.S.; Park, S.; Jeong, H.W.; Ahn, J.Y.; Choi, S.J.; Lee, H.; Choi, B.; Nam, S.K.; Sa, M.; Kwon, J.-S.; et al. Immunophenotyping of COVID-19 and Influenza Highlights the Role of Type I Interferons in Development of Severe COVID-19. Sci. Immunol. 2020, 5, eabd1554. [Google Scholar] [CrossRef]
- Amoutzias, G.D.; Nikolaidis, M.; Tryfonopoulou, E.; Chlichlia, K.; Markoulatos, P.; Oliver, S.G. The Remarkable Evolutionary Plasticity of Coronaviruses by Mutation and Recombination: Insights for the COVID-19 Pandemic and the Future Evolutionary Paths of SARS-CoV-2. Viruses 2022, 14, 78. [Google Scholar] [CrossRef]
- Min, Y.-Q.; Mo, Q.; Wang, J.; Deng, F.; Wang, H.; Ning, Y.-J. SARS-CoV-2 Nsp1: Bioinformatics, Potential Structural and Functional Features, and Implications for Drug/Vaccine Designs. Front. Microbiol. 2020, 11, 587317. [Google Scholar] [CrossRef] [PubMed]
- Afsar, M.; Narayan, R.; Akhtar, M.N.; Das, D.; Rahil, H.; Nagaraj, S.K.; Eswarappa, S.M.; Tripathi, S.; Hussain, T. Drug Targeting Nsp1-Ribosomal Complex Shows Antiviral Activity against SARS-CoV-2. eLife 2022, 11, e74877. [Google Scholar] [CrossRef] [PubMed]
- Ratia, K.; Pegan, S.; Takayama, J.; Sleeman, K.; Coughlin, M.; Baliji, S.; Chaudhuri, R.; Fu, W.; Prabhakar, B.S.; Johnson, M.E.; et al. A Noncovalent Class of Papain-like Protease/Deubiquitinase Inhibitors Blocks SARS Virus Replication. Proc. Natl. Acad. Sci. USA 2008, 105, 16119–16124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Z.; Huang, B.; Tang, J.; Liu, S.; Liu, M.; Ye, Y.; Liu, Z.; Xiong, Y.; Zhu, W.; Cao, D.; et al. The Complex Structure of GRL0617 and SARS-CoV-2 PLpro Reveals a Hot Spot for Antiviral Drug Discovery. Nat. Commun. 2021, 12, 488. [Google Scholar] [CrossRef] [PubMed]
- Pitsillou, E.; Liang, J.; Ververis, K.; Lim, K.W.; Hung, A.; Karagiannis, T.C. Identification of Small Molecule Inhibitors of the Deubiquitinating Activity of the SARS-CoV-2 Papain-Like Protease: In Silico Molecular Docking Studies and in Vitro Enzymatic Activity Assay. Front. Chem. 2020, 8, 623971. [Google Scholar] [CrossRef]

{kind=link}
| Mechanism of Inhibition | Viral Proteins | Refs. | |
|---|---|---|---|
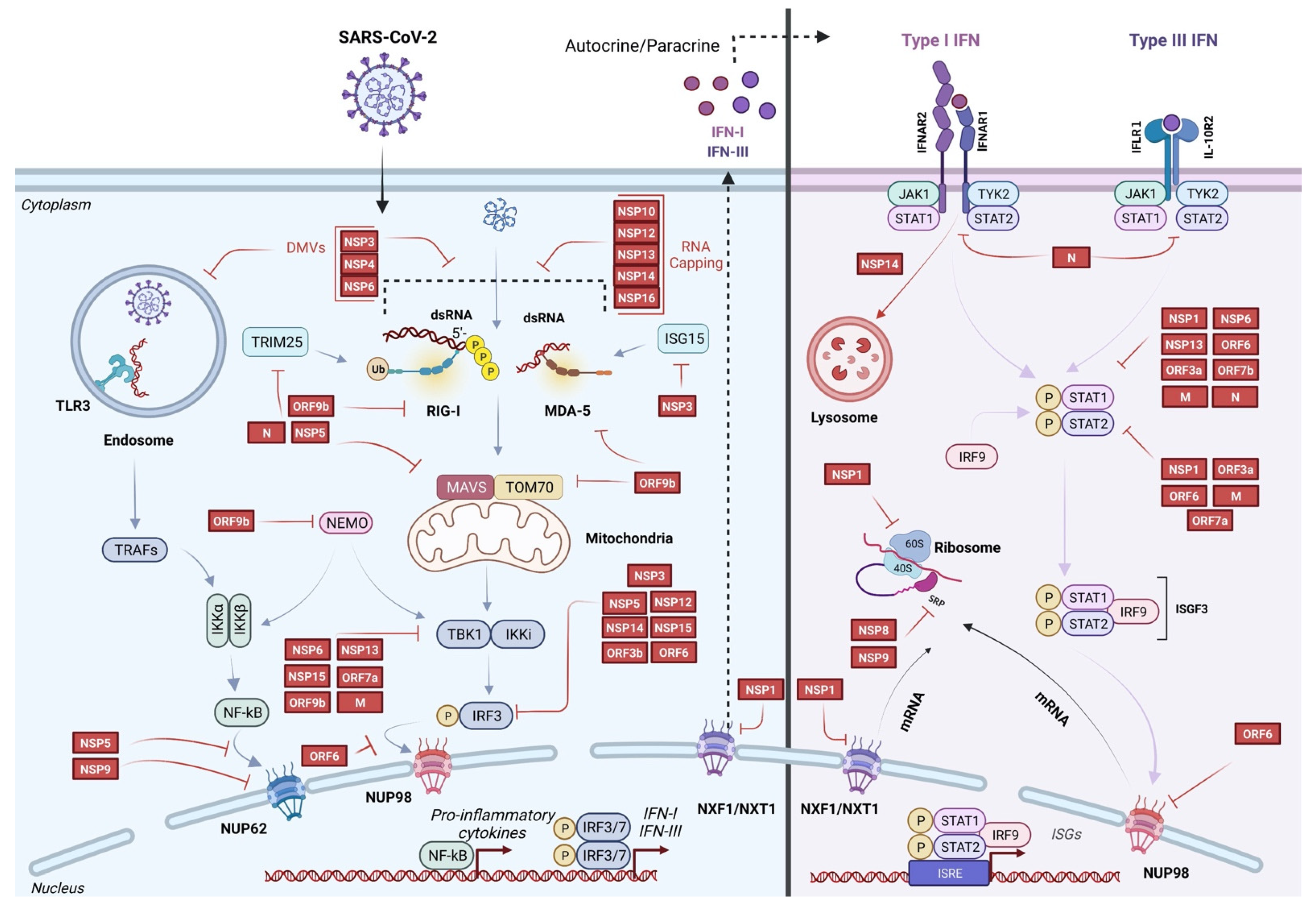
Evasion of sensing by host innate immune receptors | Formation of DMVs | NSP3 NSP4 NSP6 | [30,31] |
| Capping | NSP10 NSP12 NSP13 NSP14 NSP16 | [32,33,34] | |
| Blockage of RIG-I RNA recognition Interaction with its DExD/H helicase domain | N | [35] | |
| Cleavage of ISG15 Antagonism of ISG15-dependent MDA5 activation | NSP3 | [36,37] | |
| Cleavage of RIG-I at the last 10 N-terminal amino acids Blockage of its ability to signal through MAVS Promotion of the ubiquitination and proteosome-mediated degradation of MAVS | NSP5 | [38] | |
| Inhibition of RIG-I CARD domain activation Interaction with Trim25 | N | [39,40,41] | |
Inhibition of innate immune receptor signaling and IFNs production | Reduction of MAVS mediated IFN-β promoter activities | NSP1 NSP3 NSP5 NSP12 NSP13 NSP14 M N ORF3a ORF3b ORF6 ORF7a ORF7b ORF8 ORF9b | [42,43,44,45,46] |
| Inhibition of IRF3 phosphorylation/nuclear translocation | NSP1 NSP5 NSP6 NSP12 NSP13 NSP14 NSP15 ORF6 ORF3b | [42,43,44,49,50,51,62,63,64] | |
| Cleavage of IRF3 | NSP3 NSP5 | [62,64] | |
| Inhibition of MAVS signaling complex Interaction with TBK1 Interaction with TOM70 | NSP13 M ORF7a ORF9b | [19,44,54,55,56,57,58,59] | |
| Inhibition of NF-κB pathway Interaction with Nup69 to block p65 translocation Blockage of Nemo K63-linked polyubiquitination Cleavage of TAB1 | NSP5 NSP9 ORF9b | [60,61,62] | |
Inhibition of IFNs signaling and ISGs expression | Inhibition of STAT1/STAT2 phosphorylation | NSP1 NSP6 NSP13 M N ORF3a ORF6 ORF7a ORF7b | [44,69,70] |
| Blockage of STAT1/STAT2 nuclear translocation Interaction with the nucleopore Nup98 | ORF6 | [66,67,68] | |
| IFNAR1 lysosomal degradation | NSP14 | [65] | |
| Inhibition of ISGs | NSP3 NSP5 | [36,43,76] | |
Inhibition of protein production by targeting post-transcription and translation steps | Inhibition of pre-mRNA splicing Binding to U1 and U2 | NSP16 | [71] |
| Disruption of protein trafficking Binding to SRP complex leading to the inhibition of signal peptide recognition | NSP8 NSP9 | [71] | |
| Blockage of mRNAs export Binding to the mRNA entry channel overlapping mRNA path Interaction with export receptor heterodimer NXF1-NXT | NSP1 | [71,72,73,74,75] | |
 | ||||
|---|---|---|---|---|
| Clinical Trial Name | Type of Trial | Type of Patients | Outcomes | Refs. |
| IFN-α | ||||
| Nebulized IFN-α2b with or without Arbidol | Uncontrolled, exploratory study | 77 patients hospitalized with confirmed COVID-19 diagnosis (7 received IFN-α2b only, 46 IFN-α2b+ Arbidol) | Time to negative RT-qPCR significantly shorter in patients receiving inhaled IFN-α2b. Significant reduction in the duration of detectable virus in the upper respiratory tract. Reduced blood levels of inflammatory markers (IL-6, CRP). | [94] |
| Nebulized IFN-α2b with or without Umifenovir | Retrospective multicenter study | 446 patients with confirmed COVID-19 diagnosis (242 received IFN-α2b, 216 early and 26 late) | Nebulized IFN-α2b initiation within 5 days of admission:
| [98] |
| Inhaled IFN-α2b | Retrospective multicenter study | 1401 patients with confirmed COVID-19 diagnosis (852 received IFN-α2b) | Early administration (3–5 days after symptom onset), associated with improved clinical outcomes:
| [99] |
| PEG IFN-α2b (subcutaneous injection) in addition to standard of care [antipyretics, cough suppressants, antibiotics, steroids, vitamins, anticoagulants, hydroxychloroquine and antivirals (e.g., Remdesivir)] | Multicenter, randomized, comparator-controlled, open-label phase 3 study | 250 patients with moderate COVID-19 (120 received PEG IFN-α2b + standard of care) | Early viral clearance. Clinical status improvement. Decreased duration of supplemental oxygen. | [96] |
| IFN-α2b (subcutaneous injection) combined with Lopinavir/Ritonavir | Exploratory study | 41 patients with confirmed COVID-19 diagnosis (19 received IFN-α2b + lopinavir/ritonavir) | Early administration of IFN-α2b within 72 h following admission- resulted in shorter hospital stay, (10 days compared with late administration - after 72 h following admission). | [97] |
| IFN-β | ||||
| Nebulized IFN-β1a (inhalation) - SNG001 ClinicalTrials.gov Identifier: NCT04385095 | Double-blind randomized, placebo-controlled, phase 2 pilot study | 101 patients with confirmed COVID-19 diagnosis (50 received IFN-β1a SNG001) | Significantly greater odds of clinical improvement across the WHO Ordinal Scale for Clinical Improvement. Reduction of the odds of developing severe disease or dying. | [113] |
| Nebulized IFN-β1b (inhalation through a vibrating mesh aerogen nebulizer-Aerogen Solo) combined with Favipiravir ClinicalTrials.gov Identifier: NCT04385095 | Randomized controlled open label study | 89 adult patients hospitalized with moderate to severe COVID-19 (44 received IFN-β1b) | No significant differences in the inflammatory biomarkers at hospital discharge, in the overall lower hospital stay, transfers to the intensive care unit, neither in overall mortality. | [114] |
| IFN-β1b (subcutaneous injection) combined with Lopinavir, Ritonavir, Ribavirin | Multicenter, prospective, open label, randomized, phase 2 study | 127 patients with mild to moderate COVID-19 (86 received IFN-β1b) | Administration within 7 days of symptom onset:
| [107] |
| IFN-β1a or IFN-β1b (subcutaneous injections) combined with Lopinavir/Ritonavir COVIFERON trial ClinicalTrials.gov Identifier: NCT04343769 | Randomized, open-label, controlled study | 60 severely ill patients with positive RT-qPCR and Chest CT scans (20 patients assigned to IFN-β1a and 20 to IFN-β1b) | IFN-β1a: significant shorter time to clinical improvement. IFN-β1b: no significant difference. Lower numerically mortality both of the intervention groups (20% in the IFN-β1a group, 30% in the IFN-β1b group vs 45% in the control group) but not statically significant. | [108] |
| IFN-β1a (subcutaneous injection) in addition to the national protocol medications (Hydroxychloroquine plus Lopinavir- Ritonavir or Atazanavir-Ritonavir) Clinical Identifier: IRCT20100228003449N28 | Randomized, open-label, controlled study | 92 patients with severe COVID-19 (42 received IFN-β1a) | No change the time to reach the clinical response. Length of intensive care unit and hospital stays and duration of mechanical ventilation not statistically different. Significantly increased discharge rate on day 14. Early administration significantly reduced mortality. | [109] |
| IFN-β1a (subcutaneous injection) combined with Remdesivir ClinicalTrials.gov Identifier: NCT04492475 | Randomized, double-blind, placebo-controlled study | 969 patients hospitalized COVID-19 patients with presence of radiographic infiltrates on imaging, a peripheral oxygen saturation on room air of 94% or less, or requiring supplemental oxygen (487 received IFN-β1a) | No clinical improvement. Worse outcomes after treatment IFN-β1a in patients who required high-flow oxygen at baseline. | [111] |
| IFN-β1a (subcutaneous injection). For patients receiving high-flow oxygen, ventilation, or extra- corporeal membrane oxygenation: intravenous interferon. ClinicalTrials.gov Identifier: NCT04315948 | Randomized, double-blind, placebo-controlled study | 4127 (2063 received IFN-β1a) | No effect on hospitalized patients (based on overall mortality, initiation of ventilation, and duration of hospital stay). | [110] |
| IFN-λ | ||||
| PEG IFN-λ (subcutaneous injection) ClinicalTrials.gov Identifier: NCT04354259 | Randomized, double-blind, placebo-controlled study | 60 outpatients with COVID-19 (30 received PEG IFN-λ) | Greater decline in RT-qPCR with viral clearance by day 7. Prevent clinical deterioration and shorten duration of viral shedding. | [128] |
| PEG IFN-λ (subcutaneous injection) ClinicalTrials.gov Identifier: NCT04331899 | Randomized, double-blind, placebo-controlled phase 2 study | 120 outpatients with mild to moderate COVID-19 (30 received PEG IFN-λ) | No shortened duration of SARS-CoV-2 viral shedding. No improved of symptoms. | [129] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Znaidia, M.; Demeret, C.; van der Werf, S.; Komarova, A.V. Characterization of SARS-CoV-2 Evasion: Interferon Pathway and Therapeutic Options. Viruses 2022, 14, 1247. https://doi.org/10.3390/v14061247
Znaidia M, Demeret C, van der Werf S, Komarova AV. Characterization of SARS-CoV-2 Evasion: Interferon Pathway and Therapeutic Options. Viruses. 2022; 14(6):1247. https://doi.org/10.3390/v14061247
Chicago/Turabian StyleZnaidia, Mariem, Caroline Demeret, Sylvie van der Werf, and Anastassia V. Komarova. 2022. "Characterization of SARS-CoV-2 Evasion: Interferon Pathway and Therapeutic Options" Viruses 14, no. 6: 1247. https://doi.org/10.3390/v14061247
APA StyleZnaidia, M., Demeret, C., van der Werf, S., & Komarova, A. V. (2022). Characterization of SARS-CoV-2 Evasion: Interferon Pathway and Therapeutic Options. Viruses, 14(6), 1247. https://doi.org/10.3390/v14061247