
SARS-CoV-2 Whole-Genome Sequencing by Ion S5 Technology—Challenges, Protocol Optimization and Success Rates for Different Strains

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Genome Coverage
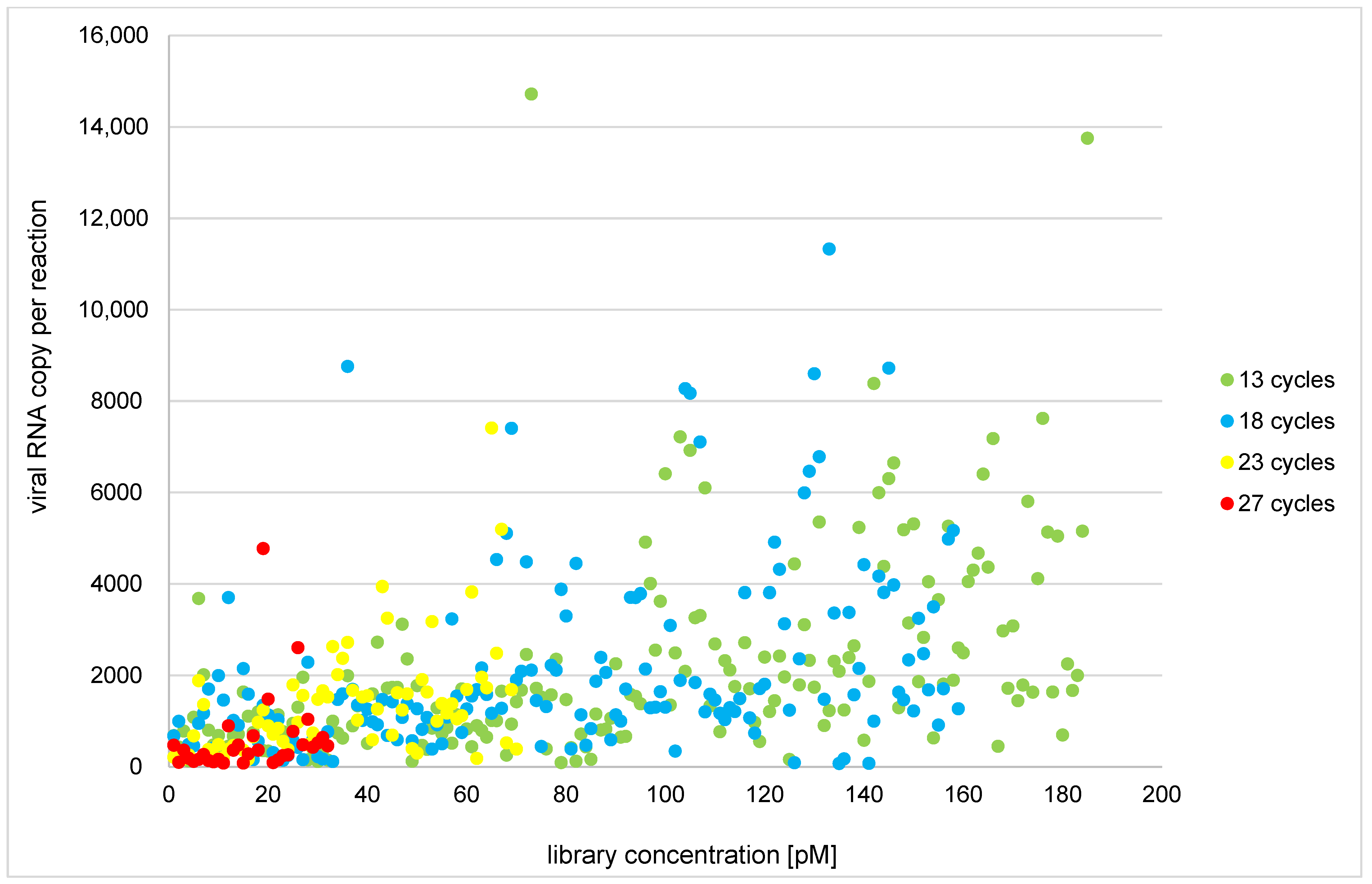
3.2. Protocol Optimization and Modification of Low-Template Sample Library Preparation
3.3. Library Reamplification
3.4. Amplicon Success Rates
3.5. Performance through Strains
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. Timeline of WHO’s Response to COVID-19. 2022. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/interactive-timeline#! (accessed on 9 March 2022).
- CDC. CDC Diagnostic Tests for COVID-19. CDC. 2022. Available online: https://www.cdc.gov/coronavirus/2019-ncov/lab/testing.html (accessed on 9 March 2022).
- FDA. In Vitro Diagnostics EUAs–Molecular Diagnostic Tests for SARS-CoV-2. FDA. 2019. Available online: https://www.fda.gov/medical-devices/coronavirus-disease-2019-covid-19-emergency-use-authorizations-medical-devices/in-vitro-diagnostics-euas-molecular-diagnostic-tests-sars-cov-2 (accessed on 9 March 2022).
- Xiao, M.; Liu, X.; Ji, J.; Li, M.; Li, J.; Yang, L.; Sun, W.; Ren, P.; Yang, G.; Zhao, J.; et al. Multiple approaches for massively parallel sequencing of SARS-CoV-2 genomes directly from clinical samples. Genome Med. 2020, 12, 57. [Google Scholar] [CrossRef]
- Munnink, B.B.O.; Nieuwenhuijse, D.F.; Stein, M.; O’Toole, A.; Haverkate, M.; Mollers, M.; Kamga, S.K.; Schapendonk, C.; Pronk, M.; Lexmond, P.; et al. Rapid SARS-CoV-2 whole-genome sequencing and analysis for informed public health decision-making in the Netherlands. Nat. Med. 2020, 26, 1405–1410. [Google Scholar] [CrossRef] [PubMed]
- Lucey, M.; Macori, G.; Mullane, N.; Sutton-Fitzpatrick, U.; Gonzalez, G.; Coughlan, S.; Purcell, A.; Fenelon, L.; Fanning, S.; Schaffer, K. Whole-genome sequencing to track severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) transmission in nosocomial outbreaks. Clin. Infect. Dis. 2021, 72, E727–E735. [Google Scholar] [CrossRef]
- Leong, L.E.; Soubrier, J.; Turra, M.; Denehy, E.; Walters, L.; Kassahn, K.; Higgins, G.; Dodd, T.; Hall, R.; D’Onise, K.; et al. Whole-genome sequencing of sars-cov-2 from quarantine hotel outbreak. Emerg. Infect. Dis. 2021, 27, 2219–2221. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, X.; Liu, Y.; Lin, J.; Shen, J.; Zhang, H.; Yin, J.; Pu, R.; Ding, Y.; Cao, G. Characterization of SARS-CoV-2 worldwide transmission based on evolutionary dynamics and specific viral mutations in the spike protein. Infect. Dis. Poverty 2021, 10, 1–15. [Google Scholar] [CrossRef]
- Bandoy, D.J.D.R.; Weimer, B.C. Analysis of SARS-CoV-2 genomic epidemiology reveals disease transmission coupled to variant emergence and allelic variation. Sci. Rep. 2021, 11, 7380. [Google Scholar] [CrossRef]
- WHO. Tracking SARS-CoV-2 Variants (who.int). 2022. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed on 9 March 2022).
- FDA. SARS-CoV-2 Viral Mutations: Impact on COVID-19 Tests. FDA. 2022. Available online: https://www.fda.gov/medical-devices/coronavirus-covid-19-and-medical-devices/sars-cov-2-viral-mutations-impact-covid-19-tests (accessed on 9 March 2022).
- Quarleri, J.; Galvan, V.; Delpino, M.V. Omicron variant of the SARS-CoV-2: A quest to define the consequences of its high mutational load. GeroScience 2022, 44, 53–56. [Google Scholar] [CrossRef]
- Chiara, M.; D’Erchia, A.M.; Gissi, C.; Manzari, C.; Parisi, A.; Resta, N.; Zambelli, F.; Picardi, E.; Pavesi, G.; Horner, D.S.; et al. Next generation sequencing of SARS-CoV-2 genomes: Challenges, applications and opportunities. Brief. Bioinform. 2021, 22, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in china. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Holmes, E.C.; Goldstein, S.A.; Rasmussen, A.L.; Robertson, D.L.; Crits-Christoph, A.; Wertheim, J.O.; Anthony, S.J.; Barclay, W.S.; Boni, M.F.; Doherty, P.C.; et al. The origins of SARS-CoV-2: A critical review. Cell 2021, 184, 4848–4856. [Google Scholar] [CrossRef]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, K.; Steininger, P.; Ziegler, R.; Steinmann, J.; Korn, K.; Ensser, A. SARS-CoV-2 samples may escape detection because of a single point mutation in the N gene. Eurosurveillance 2020, 25, 2001650. [Google Scholar] [CrossRef]
- Goletic, T.; Konjhodzic, R.; Fejzic, N.; Goletic, S.; Eterovic, T.; Softic, A.; Kustura, A.; Salihefendic, L.; Ostojic, M.; Travar, M.; et al. Phylogenetic pattern of SARS-COV-2 from COVID-19 patients from bosnia and herzegovina: Lessons learned to optimize future molecular and epidemiological approaches. Bosn. J. Basic Med. Sci. 2021, 21, 484–487. [Google Scholar] [CrossRef] [PubMed]
- Hirotsu, Y.; Omata, M. Detection of R.1 lineage severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) with spike protein W152L/E484K/G769V mutations in japan. PLoS Pathog. 2021, 17, e1009619. [Google Scholar] [CrossRef]
- Spanakis, N.; Kassela, K.; Dovrolis, N.; Bampali, M.; Gatzidou, E.; Kafasi, A.; Froukala, E.; Stavropoulou, A.; Lilakos, K.; Veletza, S.; et al. A main event and multiple introductions of SARS-CoV-2 initiated the COVID-19 epidemic in greece. J. Med. Virol. 2021, 93, 2899–2907. [Google Scholar] [CrossRef]
- Viedma, E.; Dahdouh, E.; González-Alba, J.M.; González-Bodi, S.; Martínez-García, L.; Lázaro-Perona, F.; Recio, R.; Rodríguez-Tejedor, M.; Folgueira, M.D.; Cantón, R.; et al. Genomic epidemiology of SARS-CoV-2 in madrid, spain, during the first wave of the pandemic: Fast spread and early dominance by D614G variants. Microorganisms 2021, 9, 454. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Orschler, L.; Lacknera, S. Metatranscriptomic analysis reveals SARS-CoV-2 mutations in wastewater of the frankfurt metropolitan area in southern germany. Microbiol. Resour. Announc. 2021, 10, e00280-21. [Google Scholar] [CrossRef]
- Lin, L.; Zhang, J.; Rogers, J.; Campbell, A.; Zhao, J.; Harding, D.; Sahr, F.; Liu, Y.; Wurie, I. The dynamic change of SARS-CoV-2 variants in sierra leone. Infect. Genet. Evol. 2022, 98, 105208. [Google Scholar] [CrossRef]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Cherian, S.; Potdar, V.; Vipat, V.; Ramdasi, A.; Jadhav, S.; Pawar-Patil, J.; Walimbe, A.; Patil, S.; Choudhury, M.; Shastri, J.; et al. Phylogenetic classification of the whole-genome sequences of SARS-CoV-2 from india evolutionary trends. Indian J. Med. Res. 2021, 153, 166–174. [Google Scholar] [CrossRef]
- Chen, X.; Kang, Y.; Luo, J.; Pang, K.; Xu, X.; Wu, J.; Li, X.; Jin, S. Next-generation sequencing reveals the progression of COVID-19. Front. Cell. Infect. Microbiol. 2021, 11, 632490. [Google Scholar] [CrossRef]
- Serwin, K.; Ossowski, A.; Szargut, M.; Cytacka, S.; Urbańska, A.; Majchrzak, A.; Niedźwiedź, A.; Czerska, E.; Pawińska-Matecka, A.; Gołąb, J.; et al. Molecular evolution and epidemiological characteristics of sars cov-2 in (northwestern) poland. Viruses 2021, 13, 1295. [Google Scholar] [CrossRef]
- Rachiglio, A.M.; De Sabato, L.; Roma, C.; Cennamo, M.; Fiorenza, M.; Terracciano, D.; Pasquale, R.; Bergantino, F.; Cavalcanti, E.; Botti, G.; et al. SARS-CoV-2 complete genome sequencing from the italian campania region using a highly automated next generation sequencing system. J. Transl. Med. 2021, 19, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jacot, D.; Pillonel, T.; Greub, G.; Bertelli, C. Assessment of SARS-CoV-2 genome sequencing: Quality criteria and low-frequency variants. J. Clin. Microbiol. 2021, 59, e00944-21. [Google Scholar] [CrossRef]
- Plitnick, J.; Griesemer, S.; Lasek-Nesselquist, E.; Singh, N.; Lamson, D.M.; George, K.S. Whole-genome sequencing of sars-cov-2: Assessment of the ion torrent ampliseq panel and comparison with the illumina miseq artic protocol. J. Clin. Microbiol. 2021, 59, e00649-21. [Google Scholar] [CrossRef]
- Alexandersen, S.; Chamings, A.; Bhatta, T.R. SARS-CoV-2 genomic and subgenomic RNAs in diagnostic samples are not an indicator of active replication. Nat. Commun. 2020, 11, 6059. [Google Scholar] [CrossRef]
- Wölfel, R.; Corman, V.M.; Guggemos, W.; Seilmaier, M.; Zange, S.; Müller, M.A.; Niemeyer, D.; Jones, T.C.; Vollmar, P.; Rothe, C.; et al. Virological assessment of hospitalized patients with COVID-2019. Nature 2020, 581, 465–469. [Google Scholar] [CrossRef] [Green Version]
- Telwatte, S.; Martin, H.A.; Marczak, R.; Fozouni, P.; Vallejo-Gracia, A.; Kumar, G.R.; Murray, V.; Lee, S.; Ott, M.; Wong, J.K.; et al. Novel RT-ddPCR assays for measuring the levels of subgenomic and genomic SARS-CoV-2 transcripts. Methods 2021, 201, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Mercado, N.B.; Zahn, R.; Wegmann, F.; Loos, C.; Chandrashekar, A.; Yu, J.; Liu, J.; Peter, L.; Mcmahan, K.; Tostanoski, L.H.; et al. Single-shot Ad26 vaccine protects against SARS-CoV-2 in rhesus macaques. Nature 2020, 586, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Van Doremalen, N.; Lambe, T.; Spencer, A.; Belij-Rammerstorfer, S.; Purushotham, J.N.; Port, J.R.; Avanzato, V.A.; Bushmaker, T.; Flaxman, A.; Ulaszewska, M.; et al. ChAdOx1 nCoV-19 vaccine prevents SARS-CoV-2 pneumonia in rhesus macaques. Nature 2020, 586, 578–582. [Google Scholar] [CrossRef]
- Corbett, K.S.; Flynn, B.; Foulds, K.E.; Francica, J.R.; Boyoglu-Barnum, S.; Werner, A.P.; Flach, B.; O’Connell, S.; Bock, K.W.; Minai, M.; et al. Evaluation of the mRNA-1273 vaccine against SARS-COV-2 in nonhuman primates. N. Engl. J. Med. 2020, 383, 1544–1555. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.H.; Llewelyn, A.; Brandao, R.; Chowdhary, K.; Hardisty, K.-M.; Loddo, M. SARS-CoV-2 testing and sequencing for international arrivals reveals significant cross border transmission of high risk variants into the united kingdom. EClinicalMedicine 2021, 38, 101021. [Google Scholar] [CrossRef] [PubMed]
- Alessandrini, F.; Caucci, S.; Onofri, V.; Melchionda, F.; Tagliabracci, A.; Bagnarelli, P.; Di Sante, L.; Turchi, C.; Menzo, S. Evaluation of the ion ampliseq SARS-CoV-2 research panel by massive parallel sequencing. Genes 2020, 11, 929. [Google Scholar] [CrossRef] [PubMed]
- Elwick, K.; Zeng, X.; King, J.; Budowle, B.; Hughes-Stamm, S. Comparative tolerance of two massively parallel sequencing systems to common PCR inhibitors. Int. J. Leg. Med. 2018, 132, 983–995. [Google Scholar] [CrossRef]
- Xavier, C.; de la Puente, M.; Mosquera-Miguel, A.; Freire-Aradas, A.; Kalamara, V.; Vidaki, A.; Gross, T.E.; Revoir, A.; Pośpiech, E.; Kartasińska, E.; et al. Development and validation of the VISAGE AmpliSeq basic tool to predict appearance and ancestry from DNA. Forensic Sci. Int. Genet. 2020, 48, 102336. [Google Scholar] [CrossRef] [PubMed]
- Sidstedt, M.; Steffen, C.R.; Kiesler, K.M.; Vallone, P.M.; Rådström, P.; Hedman, J. The impact of common PCR inhibitors on forensic MPS analysis. Forensic Sci. Int. Genet. 2019, 40, 182–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, K.K.; Tiong, V.; Jia-Yi, T.; Wong, J.E.; Teoh, B.T.; Abd-Jamil, J.; Johari, J.; Nor’E, S.S.; Khor, C.S.; Yaacob, C.N.; et al. Multiplex sequencing of sars-cov-2 genome directly from clinical samples using the ion personal genome machine (pgm). Trop. Biomed. 2021, 38, 283–288. [Google Scholar] [CrossRef]
- Chong, Y.M.; Sam, I.-C.; Chong, J.; Bador, M.K.; Ponnampalavanar, S.; Omar, S.F.S.; Kamarulzaman, A.; Munusamy, V.; Wong, C.K.; Jamaluddin, F.H.; et al. Sars-cov-2 lineage b.6 was the major contributor to early pandemic transmission in malaysia. PLoS Negl. Trop. Dis. 2020, 14, e0008744. [Google Scholar] [CrossRef]
- Boyle, B.; Dallaire, N.; MacKay, J. Evaluation of the impact of single nucleotide polymorphisms and primer mismatches on quantitative PCR. BMC Biotechnol. 2009, 9, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, F.C.; Torrezan, G.T.; Brianese, R.C.; Stabellini, R.; Carraro, D.M. Pitfalls in genetic testing: A case of a SNP in primer-annealing region leading to allele dropout in BRCA1. Mol. Genet. Genom. Med. 2017, 5, 443–447. [Google Scholar] [CrossRef]
- Cotten, M.; Lule Bugembe, D.; Kaleebu, P.V.T.; Phan, M. Alternate primers for whole-genome SARS-CoV-2 sequencing. Virus Evol. 2021, 7, veab006. [Google Scholar] [CrossRef] [PubMed]
- Freed, N.E.; Vlková, M.; Faisal, M.B.; Silander, O.K. Rapid and inexpensive whole-genome sequencing of SARS-CoV-2 using 1200 bp tiled amplicons and oxford nanopore rapid barcoding. Biol. Methods Protoc. 2021, 5, bpaa014. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Sequencing Batch | Number of Samples | Samples with Optimal Viral Copy Number per Reaction (≥200) | Samples that Yielded Optimal Library Conc. (≥70 pM) | Reamplified Libraries | Reamplified Libraries that Yielded Optimal Conc. (≥18 ng/mL) | Number of Fasta Files Obtained | Amplification Protocol |
|---|---|---|---|---|---|---|---|
| R1 | 80 | 78 | 65 | 9 | 8 | 80 | NORMALIZATION PROTOCOL |
| R2 | 80 | 73 | 50 | 30 | 29 | 79 | |
| SUBTOTAL 1 (R1–R2) | 160 | 151 | 115 | 39 | 37 | 159 | |
| R3 | 79 | 47 | 54 | 25 | 21 | 79 | NO-NORMALIZATION PROTOCOL |
| R4 | 79 | 78 | 78 | 1 | 0 | 78 | |
| R5 | 77 | 51 | 75 | 2 | 0 | 77 | |
| R6 | 77 | 76 | 77 | 0 | - | 77 | |
| R7 | 81 | 81 | 81 | 0 | - | 81 | |
| R8 | 81 | 81 | 81 | 0 | - | 81 | |
| SUBTOTAL 2 (R3–R8) | 474 | 414 | 446 | 28 | 21 | 473 | |
| TOTAL | 634 | 565 | 561 | 67 | 58 | 632 |
| N GENE CT | VIRAL RNA COPY PER REACTION | LIBRARY CONC. (pM) (TARGET 70 pM) | REAMPLI-FIED LIBRARY CONC. (ng/mL) (TARGET 18 ng/mL) | READS | ON TARGET | MEAN DEPTH | UNIFORMITY | ||
|---|---|---|---|---|---|---|---|---|---|
| LOW-TEMPLATE SAMPLES SUBJECTED TO RNA CONCENTRATION | MEAN | 30.1 | 99.3 | 340.1 | 73.0 | 525,759.0 | 72.06% | 2380.7 | 42.96% |
| MEDIAN | 29.0 | 78.0 | 86.6 | 31.6 | 347,048.0 | 84.94% | 503.9 | 38.67% | |
| SD | 2.0 | 50.9 | 721.1 | 106.3 | 560,685.2 | 26.48% | 3126.2 | 30.29% | |
| LOW-TEMPLATE SAMPLES, NOT SUBJECTED TO RNA CONCENTRATION | MEAN | 29.0 | 104.0 | 100.2 | 105.9 | 566,962.3 | 74.48% | 2174.1 | 43.41% |
| MEDIAN | 29.0 | 78.0 | 74.7 | 91.4 | 580,470.0 | 77.98% | 718.1 | 38.91% | |
| SD | 1.1 | 48.6 | 90.3 | 68.5 | 440,854.7 | 16.89% | 2370.6 | 26.86% | |
| N GENE CT | VIRAL RNA COPY PER REACTION | LIBRARY CONC. (pM) (TARGET 70 pM) | REAMPLIFIED LIBRARY CONC. (ng/mL) (TARGET 18 ng/mL) | READS | ON TARGET | MEAN DEPTH | UNIFORMITY | ||
|---|---|---|---|---|---|---|---|---|---|
| SUMMARIZED | OVERALL | 20.7 | 517,043.1 | 1432.7 | 72.0 | 1,096,133.6 | 94.74% | 6866.4 | 80.66% |
| R1–R2 | 19.5 | 103,894.7 | 283.6 | 72.4 | 1,053,337.7 | 96.28% | 6400.0 | 73.60% | |
| R3–R8 | 21.8 | 685,312.1 | 1817.4 | 71.5 | 1,110,459.0 | 93.97% | 7023.5 | 84.18% | |
| OPTIMAL CONC. | OVERALL | 20.4 | 578,483.9 | 1616.2 | - | 1,209,977.6 | 97.18% | 7618.0 | 88.93% |
| R1–R2 | 18.7 | 122,172.9 | 379.4 | - | 1,348,146.2 | 97.64% | 8315.9 | 85.08% | |
| R3–R8 | 21.5 | 713,803.7 | 1919.5 | - | 1,176,098.3 | 97.01% | 7446.2 | 90.34% | |
| OPTIMAL INITIAL RNA INPUT | OVERALL | 19.7 | 574,396.9 | 1570.4 | 66.3 | 1,165,060.8 | 98.38% | 7411.8 | 86.80% |
| R1–R2 | 19.0 | 108,337.3 | 292.7 | 66.3 | 1,078,694.5 | 97.28% | 6639.7 | 74.95% | |
| R3–R8 | 20.3 | 775,134.2 | 2036.4 | - | 1,196,561.5 | 99.02% | 7694.9 | 93.68% | |
| OPTIMAL CONC. AND INITIAL RNA INPUT | OVERALL | 20.1 | 737,264.0 | 1772.5 | - | 1,213,934.0 | 98.71% | 7750.6 | 91.40% |
| R1–R2 | 19.5 | 124,233.1 | 398.5 | - | 1,311,516.7 | 97.72% | 8094.1 | 84.08% | |
| R3–R8 | 20.3 | 924,833.2 | 2044.0 | - | 1,194,652.6 | 99.03% | 7682.4 | 93.70% | |
| REAMPLIFIED | OVERALL | 23.1 | 33,212.4 | 29.4 | 72.0 | 167,032.7 | 80.00% | 723.8 | 32.84% |
| R1–R2 | 22.3 | 45,257.7 | 30.9 | 72.1 | 248,339.7 | 90.58% | 1142.5 | 37.93% | |
| R3–R8 | 31.0 | 87.8 | 27.4 | 72.0 | 53,783.7 | 64.71% | 119.1 | 25.48% | |
| REAMPLIFIED WITH OPTIMAL CONC. | OVERALL | 22.6 | 35,579.1 | 29.5 | 81.5 | 184,595.0 | 79.81% | 801.0 | 35.36% |
| R1–R2 | 22.3 | 45,257.7 | 30.9 | 72.1 | 248,339.7 | 90.58% | 1142.5 | 37.93% | |
| R3–R8 | 33.0 | 91.0 | 26.5 | 100.9 | 53,750.7 | 57.71% | 100.0 | 30.07% |
| LINEAGE | NO OF SAMPLES | TARGET REGION | RANGE | RANGE LOST IF AMPLICON FAILS | MEAN % READS WITHIN ALL SAMPLES | CHANGE | POSITION OF VARIANT IN RELATION TO AMPLICON RANGE 1 | VARIANT | % READS | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| B.1.1.306 | N = 1 | r1_1.1.592753 | 316 | 535 | 484 | 512 | 0.854% | 313C>T | 3U | ||
| B.1.1.44 | N = 1 | r1_1.10.67083 | 9179 | 9390 | 9199 | 9251 | 0.199% | 9166C>A | 13U | T | 0.005% |
| B.1.389 | N = 1 | r1_1.10.711902 | 9432 | 9643 | 9477 | 9561 | 0.377% | 9430C>T | 2U | T | 0.004% |
| T | 0.002% | ||||||||||
| B.1.1.317 | N = 1 | r1_1.11.376074 | 9857 | 10,071 | 9906 | 9952 | 0.388% | 9857C>T | 0U | T | 0.008% |
| AY.9 | N = 2 | r1_1.12.1117806 | 11,523 | 11,730 | 11,634 | 11,684 | 0.344% | 11514C>T | 9U | T | 0.002% |
| B.1.160 | N = 2 | r1_1.12.539895 | 11,260 | 11,477 | 11,366 | 11,410 | 0.434% | 11497C>T | 20D | T | 0.082% |
| B.1.1.306 | N = 1 | r1_1.13.620498 | 12,563 | 12,790 | 12,711 | 12,772 | 0.630% | 12805T>C | 15D | ||
| AY.1 | N = 2 | r1_1.15.1421280 | 14,410 | 14,550 | 14,473 | 14,514 | 0.007% | 14408C>T | 2U | ||
| AY.9 | N = 2 | ||||||||||
| B.1 | N = 9 | ||||||||||
| B.1.1 | N = 22 | ||||||||||
| B.1.389 | N = 1 | r1_1.16.1212393 | 15,560 | 15,741 | 15,583 | 15,624 | 0.301% | 15543G>T | 17U | ||
| B.1.258 | N = 12 | r1_1.16.534874 | 15,387 | 15,582 | 15,522 | 15,559 | 0.879% | 15598G>A | 16D | ||
| B.1.389 | N = 1 | r1_1.21.272458 | 20,380 | 20,604 | 20,457 | 20,497 | 0.372% | 20622A>T 20623G>T 20624A>T | 18D | ||
| B.1.1.398 | N = 4 | r1_1.22.1029456 | 21,187 | 21,255 | 21,199 | 21,245 | 0.409% | 21178C>T | 9U | ||
| B.1.2 | N = 5 | r1_1.22.906171 | 21,309 | 21,531 | 21,346 | 21,450 | 0.351% | 21304C>T | 5U | A | 0.002% |
| A | 0.004% | ||||||||||
| AY.9 | N = 2 | r1_1.23.1186794 | 22,246 | 22,446 | 22,320 | 22,367 | 0.407% | 22227C>T | 19U | T | 0.006% |
| B.1.177 | N = 9 | T | 0.004% | ||||||||
| B.1.177.8 | N = 1 | T | 0.003% | ||||||||
| T | 0.009% | ||||||||||
| T | 0.006% | ||||||||||
| AY.1 | N = 2 | r1_1.23.127614 | 21,757 | 21,973 | 21,803 | 21,846 | 0.095% | 21987G>A | 14D | ||
| AY.9 | N = 2 | ||||||||||
| AY.1 | N = 2 | r1_1.23.86525 | 21,847 | 22,058 | 21,974 | 22,024 | 0.325% | 21846C>T | 1U | ||
| B.1.617.2 | N = 1 | ||||||||||
| B.1.389 | N = 1 | r1_1.24.394902 | 23,580 | 23,724 | 23,643 | 23,682 | 0.601% | 23730C>T | 6D | ||
| AY.9 | N = 2 | r1_1.24.626090 | 23,008 | 23,224 | 23,049 | 23,088 | 0.380% | 22995C>A | 13U | A | 0.022% |
| AY.1 | N = 2 | ||||||||||
| B.1.221 | N = 32 | r1_1.26.1209362 | 25,666 | 25,887 | 25,797 | 25,844 | 0.249% | 25906G>C | 19D | T | 0.008% |
| C/G (1382/3601) | 0.284% | ||||||||||
| B.1.1.121 | N = 3 | r1_1.27.410513 | 26,651 | 26,847 | 26,801 | 26,838 | 0.424% | 26645C>T | 6U | ||
| AY.9 | N = 2 | r1_1.3.760885 | 2398 | 2616 | 2300 | 2463 | 0.263% | 2388C>T | 10U | ||
| B.1.258 | N = 12 | r1_1.30.1041188 | 29,498 | 29,727 | 29,530 | 29,592 | 0.328% | 29734G>C | 7D | ||
| AY.1 | N = 2 | 29742G>T | 15D | T | 0.013% | ||||||
| AY.9 | N = 2 | T | 0.029% | ||||||||
| T | 0.100% | ||||||||||
| B.1.1.159 | N = 2 | r1_1.5.75163 | 3891 | 4110 | 3973 | 4018 | 0.240% | 4114T>C | 4D | ||
| B.1.1.306 | N = 1 | 3879–3899del | 12U | ||||||||
| B.1.1.44 | N = 1 | r1_1.6.1402513 | 4920 | 5129 | 5028 | 5073 | 0.385% | 5147C>T | 18D | T | 0.024% |
| AY.9 | N = 2 | r1_1.6.888565 | 5587 | 5810 | 5671 | 5716 | 0.481% | 5584A>G | 3U | ||
| B.1.1.317 | N = 1 | r1_1.7.488632 | 6293 | 6512 | 6339 | 6379 | 0.561% | 6536G>A | 24D | ||
| B.1.177 | N = 9 | 6286C>T | 7U | ||||||||
| B.1.177.8 | N = 1 | ||||||||||
| B.1.1.44 | N = 1 | r1_1.8.592180 | 6804 | 7017 | 6830 | 6882 | 0.210% | - | |||
| B.1.1.159 | N = 2 | r1_1.8.816048 | 7149 | 7360 | 7284 | 7331 | 0.149% | 7379G>A | 19D | ||
| LINEAGE | NO OF SAMPLES | TARGET REGION | RANGE | RANGE LOST IF AMPLICON FAILS | MEAN % READS WITHIN ALL SAMPLES | CHANGE | POSITION OF VARIANT IN RELATION TO AMPLICON RANGE 1 | ||
|---|---|---|---|---|---|---|---|---|---|
| B.1.1.529 | N = 5 | r1_1.11.528369 | 10,453 | 10,679 | 10,603 | 10,655 | 0.197% | 10449C>A | 4U |
| r1_1.15.1421280 | 14,410 | 14,550 | 14,473 | 14,514 | 0.007% | 14408C>T | 2U | ||
| r1_1.23.127614 | 21,757 | 21,973 | 21,803 | 21,846 | 0.095% | 21987G>A | 14D | ||
| r1_1.23.474025 | 22,494 | 22,685 | 22,582 | 22,622 | 0.436% | 22686C>T | 1D | ||
| r1_1.24.942468 | 23,089 | 23,290 | 23,225 | 23,268 | 0.256% | 23075T>C | 14U | ||
| r1_1.26.781963 | 25,372 | 25,566 | 25,478 | 25,511 | 0.785% | 25584C>T | 18D | ||
| r1_1.27.993816 | 26,588 | 26,800 | 26,606 | 26,650 | 0.332% | 26577C>G | 11U | ||
| r1_1.29.497787 | 28,374 | 28,606 | 28,463 | 28,512 | 0.521% | 28,362–28,370del * | 4U | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szargut, M.; Cytacka, S.; Serwin, K.; Urbańska, A.; Gastineau, R.; Parczewski, M.; Ossowski, A. SARS-CoV-2 Whole-Genome Sequencing by Ion S5 Technology—Challenges, Protocol Optimization and Success Rates for Different Strains. Viruses 2022, 14, 1230. https://doi.org/10.3390/v14061230
Szargut M, Cytacka S, Serwin K, Urbańska A, Gastineau R, Parczewski M, Ossowski A. SARS-CoV-2 Whole-Genome Sequencing by Ion S5 Technology—Challenges, Protocol Optimization and Success Rates for Different Strains. Viruses. 2022; 14(6):1230. https://doi.org/10.3390/v14061230
Chicago/Turabian StyleSzargut, Maria, Sandra Cytacka, Karol Serwin, Anna Urbańska, Romain Gastineau, Miłosz Parczewski, and Andrzej Ossowski. 2022. "SARS-CoV-2 Whole-Genome Sequencing by Ion S5 Technology—Challenges, Protocol Optimization and Success Rates for Different Strains" Viruses 14, no. 6: 1230. https://doi.org/10.3390/v14061230
APA StyleSzargut, M., Cytacka, S., Serwin, K., Urbańska, A., Gastineau, R., Parczewski, M., & Ossowski, A. (2022). SARS-CoV-2 Whole-Genome Sequencing by Ion S5 Technology—Challenges, Protocol Optimization and Success Rates for Different Strains. Viruses, 14(6), 1230. https://doi.org/10.3390/v14061230