
CAVES: A Novel Tool for Comparative Analysis of Variant Epitope Sequences


Abstract
:1. Introduction
2. Materials and Methods
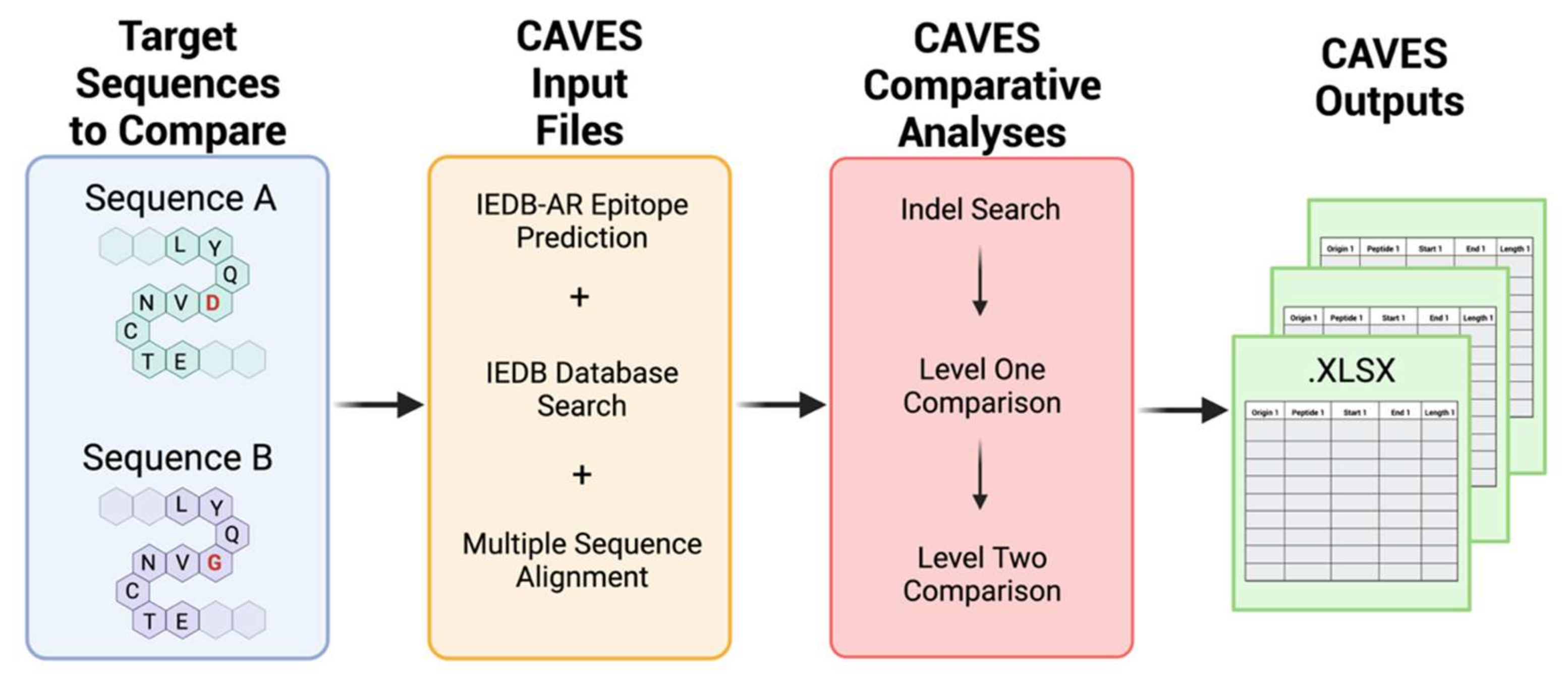
2.1. CAVES Concepts of Design
2.1.1. Compatibility with the IEDB
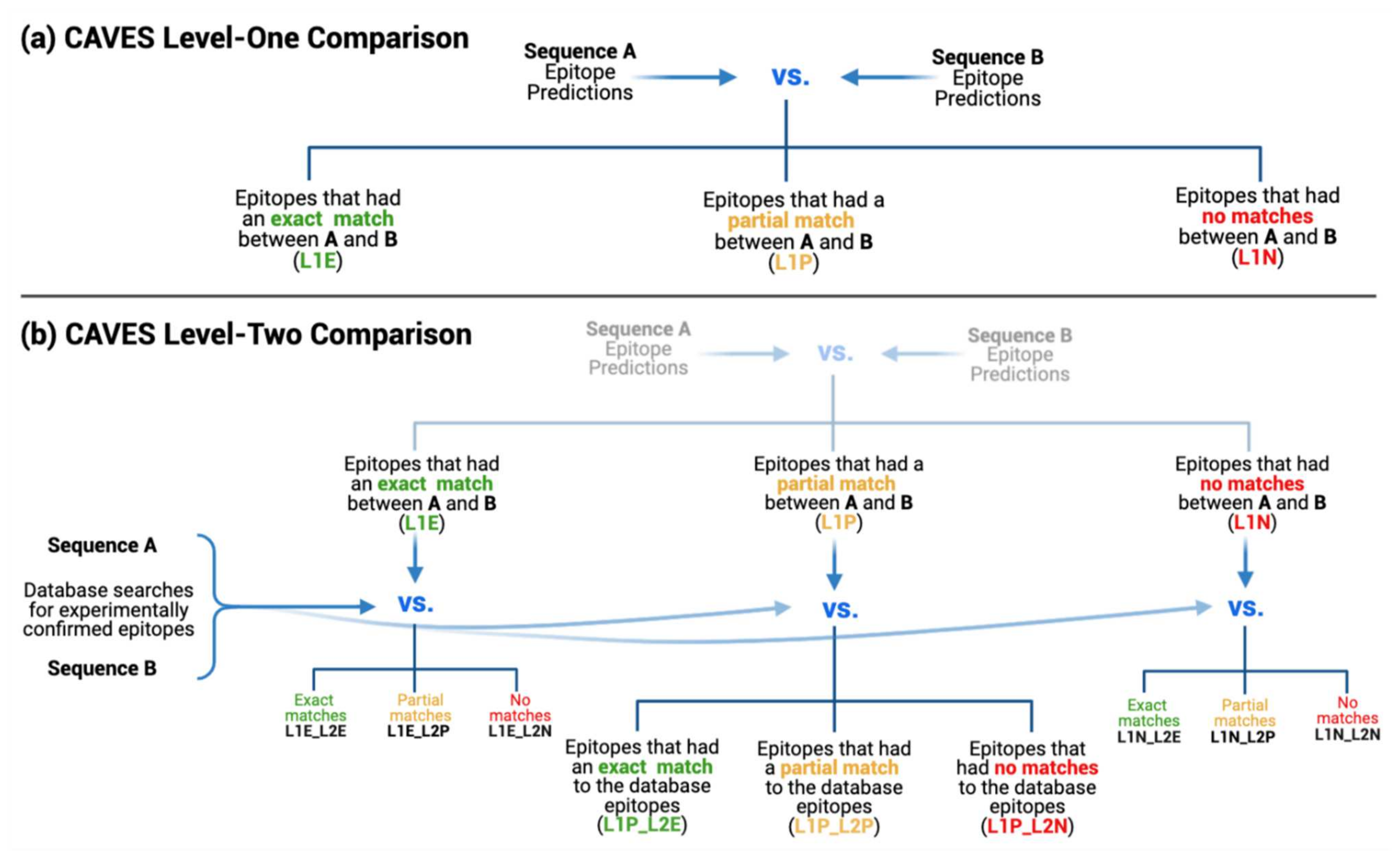
2.1.2. Two-Level Comparative Analysis
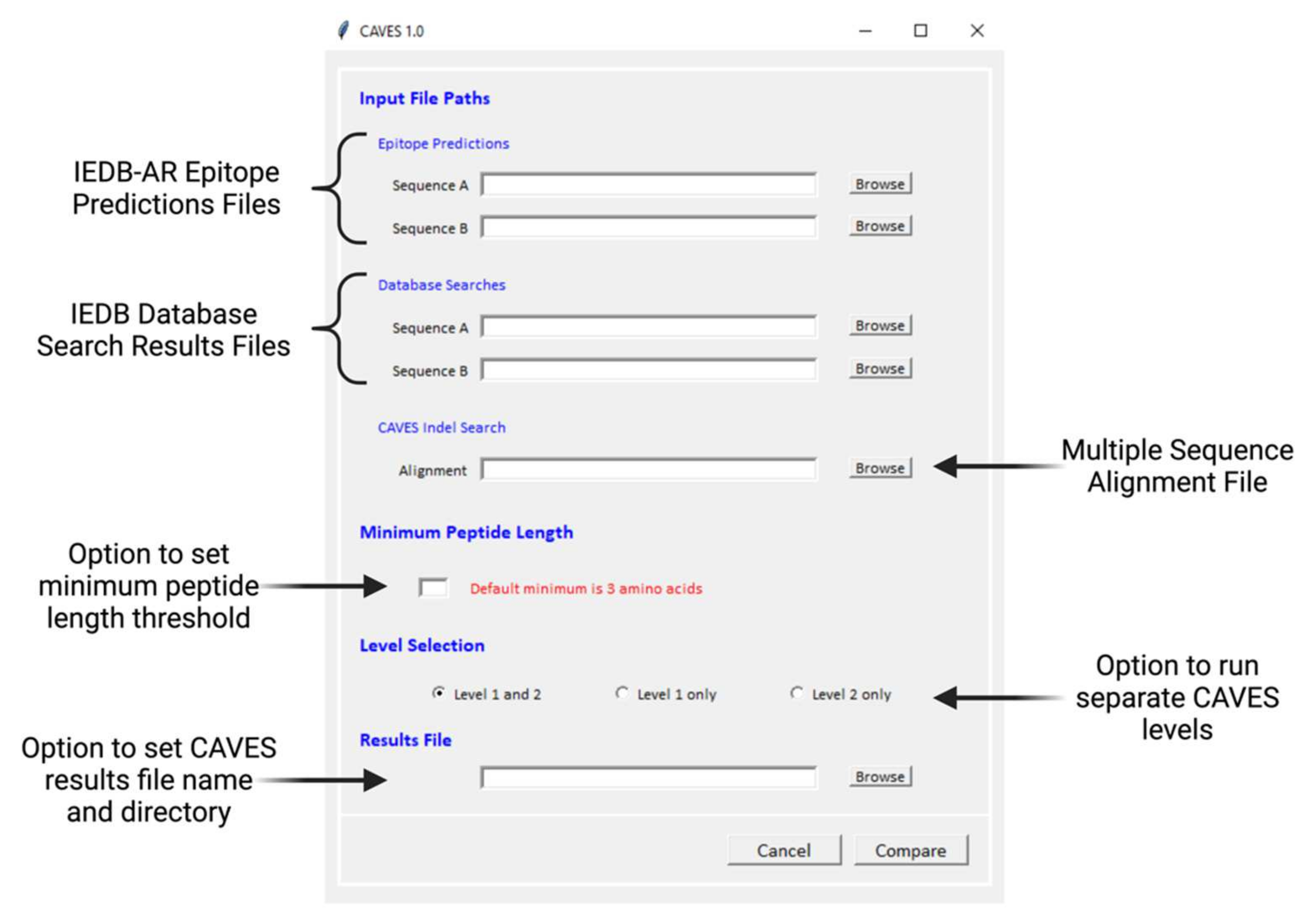
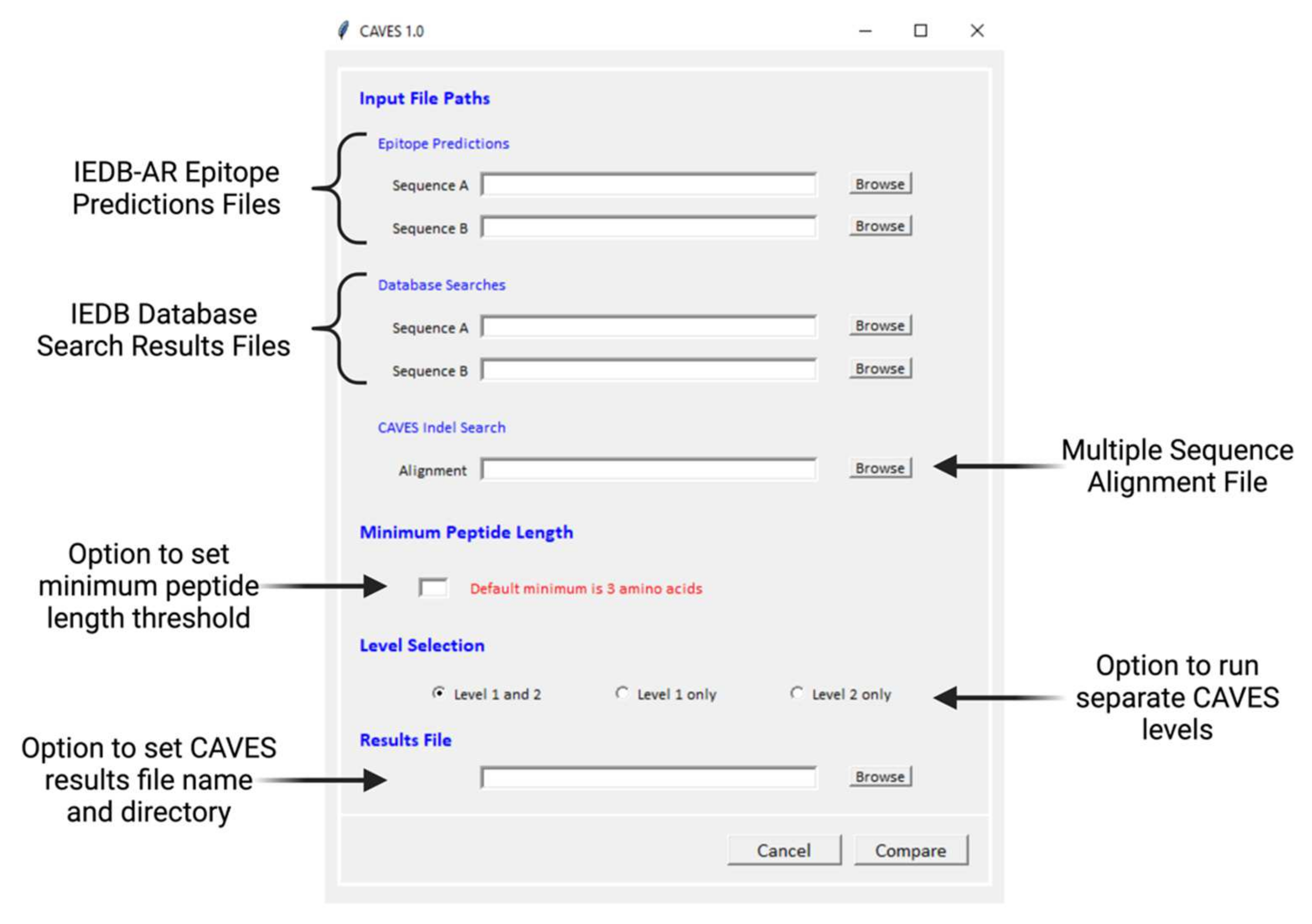
2.1.3. Optional Parameter Settings
2.1.4. CAVES Download and GUI
2.1.5. CAVES Output
2.2. CAVES Comparative Process
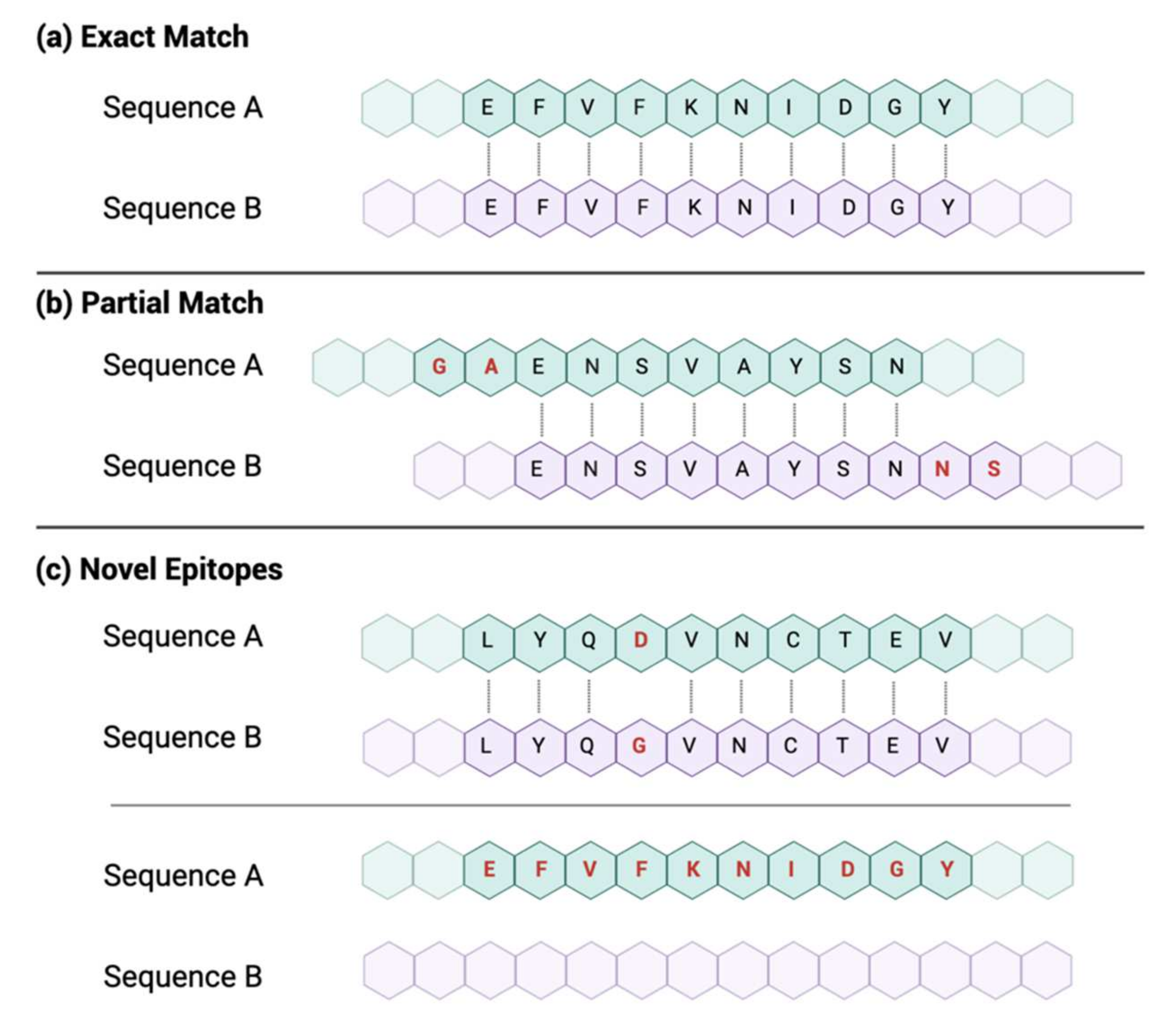
2.2.1. Matching Criteria
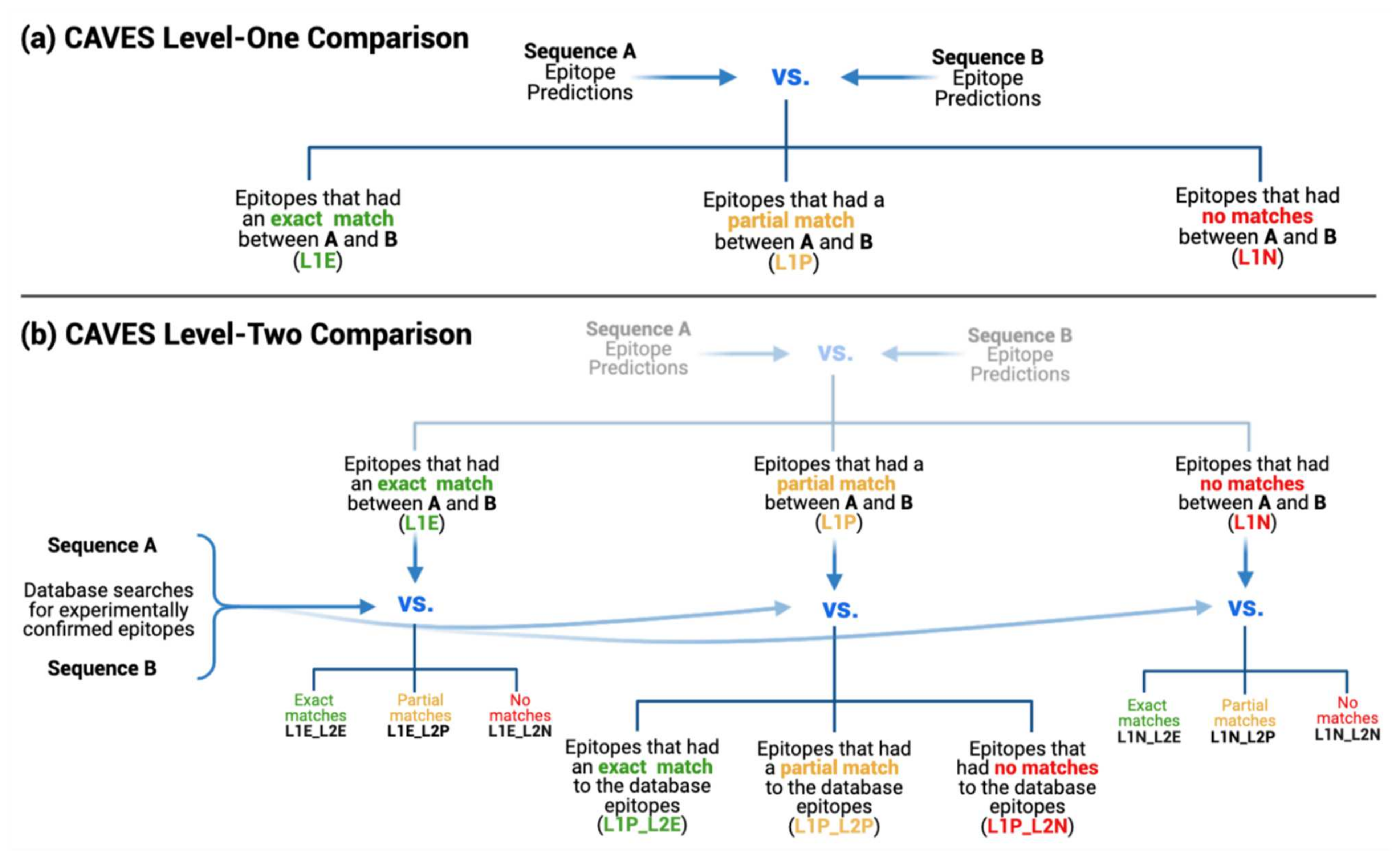
2.2.2. Level-One Comparison
2.2.3. Level-Two Comparison
2.3. Sample Dataset
2.3.1. Sequence Downloads and Manipulation
2.3.2. IEDB Epitope Prediction
2.3.3. IEDB Database Search
2.3.4. Multiple Sequence Alignment and CAVES Analysis
2.3.5. Additional HIV-1 CAVES Analysis
3. Results
3.1. Epitope Prediction and the IEDB Database Searches
3.2. CAVES L1
3.3. CAVES L2
3.4. Additional HIV-1 CAVES Analysis Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yurina, V. Coronavirus epitope prediction from highly conserved region of spike protein. Clin. Exp. Vaccine Res. 2020, 9, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Dhanda, S.K.; Mahajan, S.; Paul, S.; Yan, Z.; Kim, H.; Jespersen, M.C.; Jurtz, V.; Andreatta, M.; Greenbaum, J.A.; Marcatili, P.; et al. IEDB-AR: Immune epitope database-analysis resource in 2019. Nucleic Acids Res. 2019, 47, W502–W506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, S.; Sidney, J.; Sette, A.; Peters, B. TepiTool: A pipeline for computational prediction of T cell epitope candidates. Curr. Protoc. Immunol. 2016, 114, 18.19.1–18.19.24. [Google Scholar] [CrossRef] [PubMed]
- Martini, S.; Nielsen, M.; Peters, B.; Sette, A. The Immune Epitope Database and Analysis Resource Program 2003-2018: Reflections and outlook. Immunogenetics 2020, 72, 57–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vita, R.; Overton, J.A.; Greenbaum, J.A.; Ponomarenko, J.; Clark, J.D.; Cantrell, J.R.; Wheeler, D.K.; Gabbard, J.L.; Hix, D.; Sette, A.; et al. The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 2014, 43, D405–D412. [Google Scholar] [CrossRef]
- Jespersen, M.C.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-2.0: Improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017, 45, W24–W29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2021, 50, D20–D26. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Karosiene, E.; Rasmussen, M.; Blicher, T.; Lund, O.; Buus, S.; Nielsen, M. NetMHCIIpan-3.0, a common pan-specific MHC class II prediction method including all three human MHC class II isotypes, HLA-DR, HLA-DP and HLA-DQ. Immunogenetics 2013, 65, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.; Lundegaard, C.; Blicher, T.; Peters, B.; Sette, A.; Justesen, S.; Buus, S.; Lund, O. Quantitative Predictions of Peptide Binding to Any HLA-DR Molecule of Known Sequence: NetMHCIIpan. PLoS Comput. Biol. 2008, 4, e1000107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vita, R.; Mahajan, S.; Overton, J.A.; Dhanda, S.K.; Martini, S.; Cantrell, J.R.; Wheeler, D.K.; Sette, A.; Peters, B. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 2019, 47, D339–D343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foley, B.T.; Korber, B.T.M.; Leitner, T.K.; Apetrei, C.; Hahn, B.; Mizrachi, I.; Mullins, J.; Rambaut, A.; Wolinsky, S. HIV Sequence Compendium 2018; Theoretical Biology and Biophysics Group, Los Alamos National Lab.: Los Alamos, NM, USA, 2018. [Google Scholar]
- Yusim, K.; David-Fung, E.-S.; Korber, B.T.M.; Brander, C.; Barouch, D.; de Boer, R.; Haynes, B.F.; Koup, R.; Moore, J.P.; Walker, B.D.; et al. HIV Molecular Immunology 2020; Theoretical Biology and Biophysics Group, Los Alamos National Lab.: Los Alamos, NM, USA, 2020. [Google Scholar]
- Huang, Y.; Yang, C.; Xu, X.; Xu, W.; Liu, S. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, Á.; Kraemer, M.U.G.; Hill, V.; Pybus, O.G.; Watts, A.; Bogoch, I.I.; Khan, K.; Messina, J.P.; Tegally, H.; Lessells, R.R.; et al. Tracking the international spread of SARS-CoV-2 lineages B.1.1.7 and B.1.351/501Y-V2. Wellcome Open Res. 2021, 6, 121. [Google Scholar] [CrossRef]
- Galloway, S.E.; Paul, P.; MacCannell, D.R.; Johansson, M.A.; Brooks, J.T.; MacNeil, A.; Slayton, R.B.; Tong, S.; Silk, B.J.; Armstrong, G.L.; et al. Emergence of SARS-CoV-2 B.1.1.7 Lineage—United States, 29 December 2020–12 January 2021; Centers for Disease Control MMWR Office: Atlanta, GA, USA, 2021; Volume 70. [Google Scholar]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohail, M.S.; Ahmed, S.F.; Quadeer, A.A.; Mckay, M.R. In silico T cell epitope identification for SARS-CoV-2: Progress and perspectives. Adv. Drug Deliv. Rev. 2021, 171, 29–47. [Google Scholar] [CrossRef]
- Lon, J.R.; Bai, Y.; Zhong, B.; Cai, F.; Du, H. Prediction and evolution of B cell epitopes of surface protein in SARS-CoV-2. Virol. J. 2020, 17, 165. [Google Scholar] [CrossRef]
- Wang, X.; Sun, Q.; Ye, Z.; Hua, Y.; Shao, N.; Du, Y.; Zhang, Q.; Wan, C. Computational approach for predicting the conserved B-cell epitopes of hemagglutinin H7 subtype influenza virus. Exp. Ther. Med. 2016, 12, 2439–2446. [Google Scholar] [CrossRef] [Green Version]
- Fleri, W.; Paul, S.; Dhanda, S.K.; Mahajan, S.; Xu, X.; Peters, B.; Sette, A. The Immune Epitope Database and Analysis Resource in Epitope Discovery and Synthetic Vaccine Design. Front. Immunol. 2017, 8, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CAVES Results Category a | Number of Epitopes b | Number Derived from Reference Sequence | Number Derived from Alpha VOC Sequence |
|---|---|---|---|
| L1E | 93 | ||
| L1P | 159 | ||
| L1N | 25 | 12 | 13 |
| L1E_L2E | 82 | 42 | 40 |
| L1E_L2P | 444 | 229 | 215 |
| L1E_L2N | 15 | 7 | 8 |
| L1P_L2E | 85 | 44 | 41 |
| L1P_L2P | 462 | 229 | 233 |
| L1P_L2N | 16 | 7 | 9 |
| L1N_L2E | 6 | 6 | 0 |
| L1N_L2P | 49 | 28 | 21 |
| L1N_L2N | 3 | 0 | 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, K.; Lowey, C.; Sandstrom, P.; Ji, H. CAVES: A Novel Tool for Comparative Analysis of Variant Epitope Sequences. Viruses 2022, 14, 1152. https://doi.org/10.3390/v14061152
Li K, Lowey C, Sandstrom P, Ji H. CAVES: A Novel Tool for Comparative Analysis of Variant Epitope Sequences. Viruses. 2022; 14(6):1152. https://doi.org/10.3390/v14061152
Chicago/Turabian StyleLi, Katherine, Connor Lowey, Paul Sandstrom, and Hezhao Ji. 2022. "CAVES: A Novel Tool for Comparative Analysis of Variant Epitope Sequences" Viruses 14, no. 6: 1152. https://doi.org/10.3390/v14061152
APA StyleLi, K., Lowey, C., Sandstrom, P., & Ji, H. (2022). CAVES: A Novel Tool for Comparative Analysis of Variant Epitope Sequences. Viruses, 14(6), 1152. https://doi.org/10.3390/v14061152