
Discovery of a Novel Jingmenvirus in Australian Sugarcane Soldier Fly (Inopus flavus) Larvae




Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and RNA Extraction
2.2. Transcriptome Data Analysis and Virus Discovery
2.3. Phylogenetic Analysis
2.4. In Vitro Isolation Attempts
2.5. Viral Derived Small RNA Analysis
3. Results
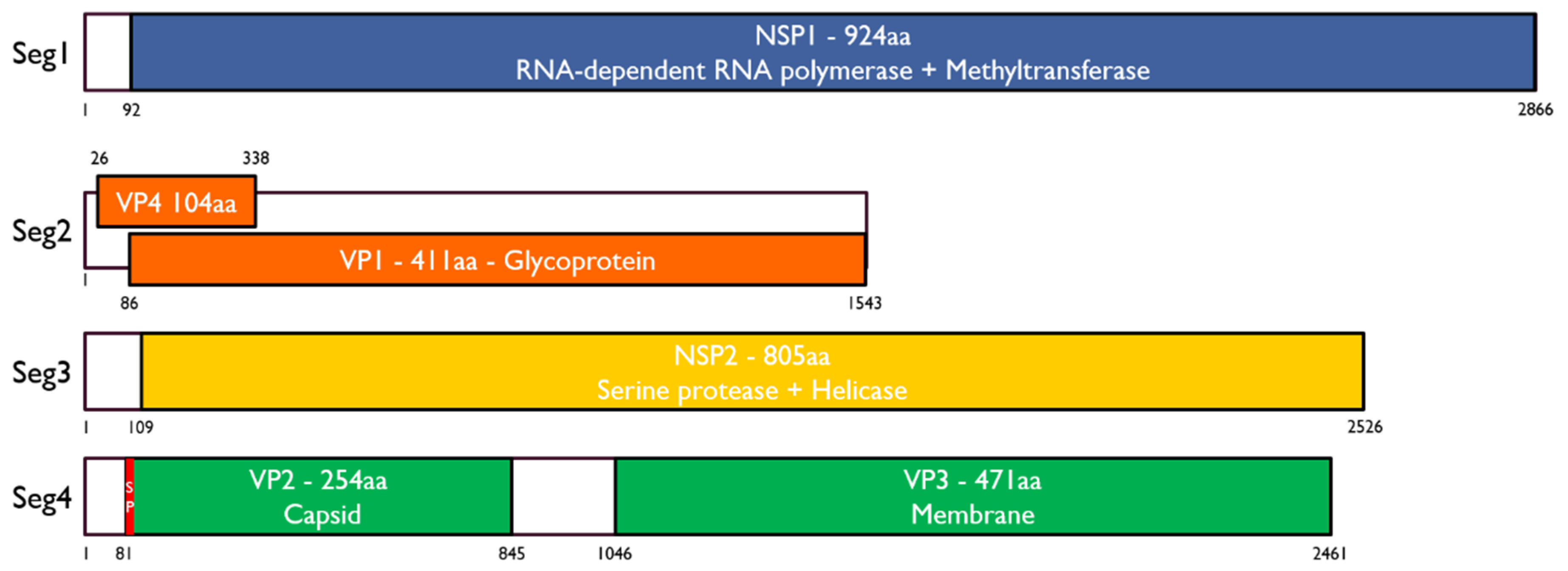
3.1. Identification of Inopus Flavus Jingmenvirus 1
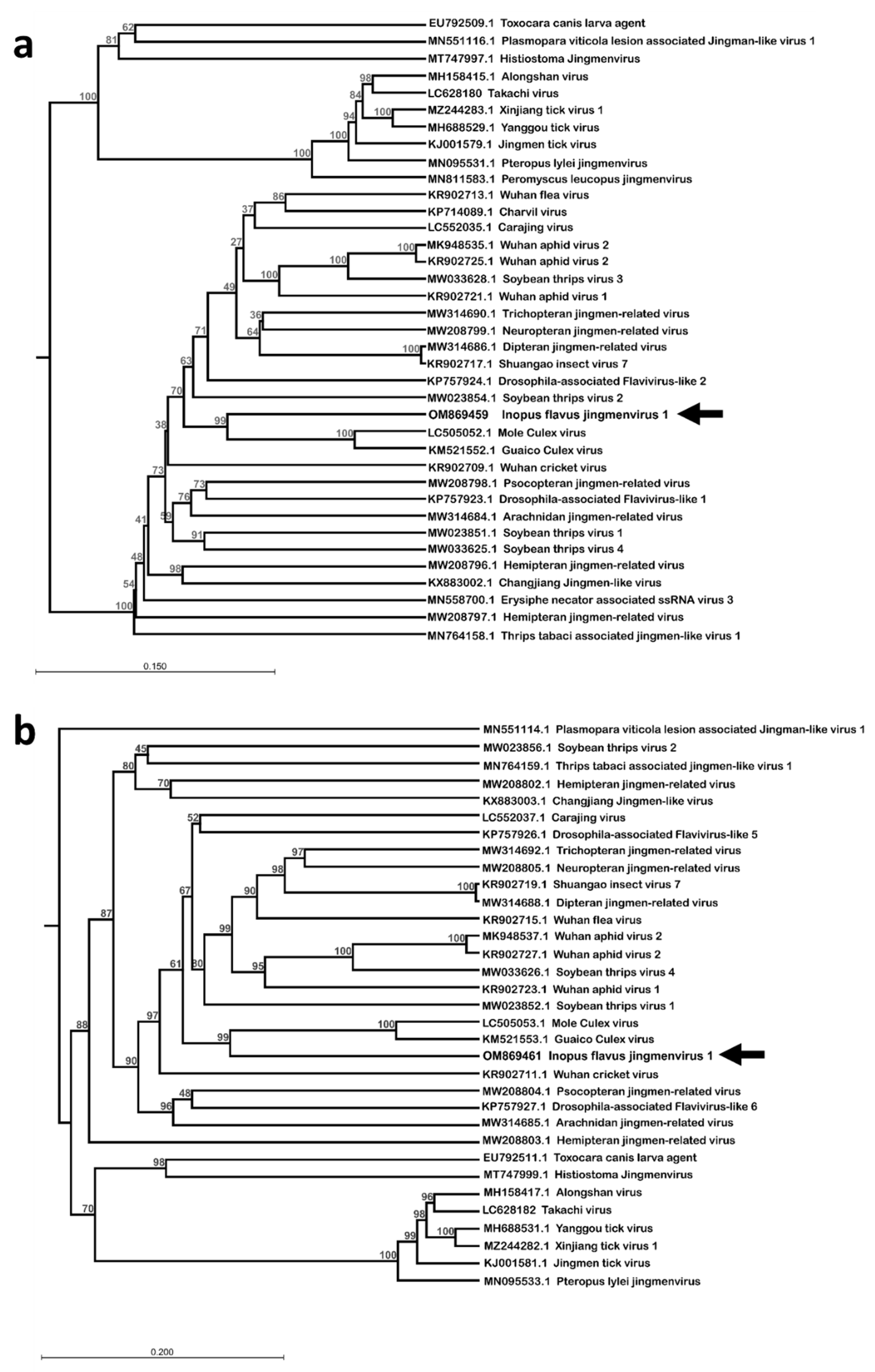
3.2. Phylogenetic Analysis
3.3. In Vitro Virus Isolation
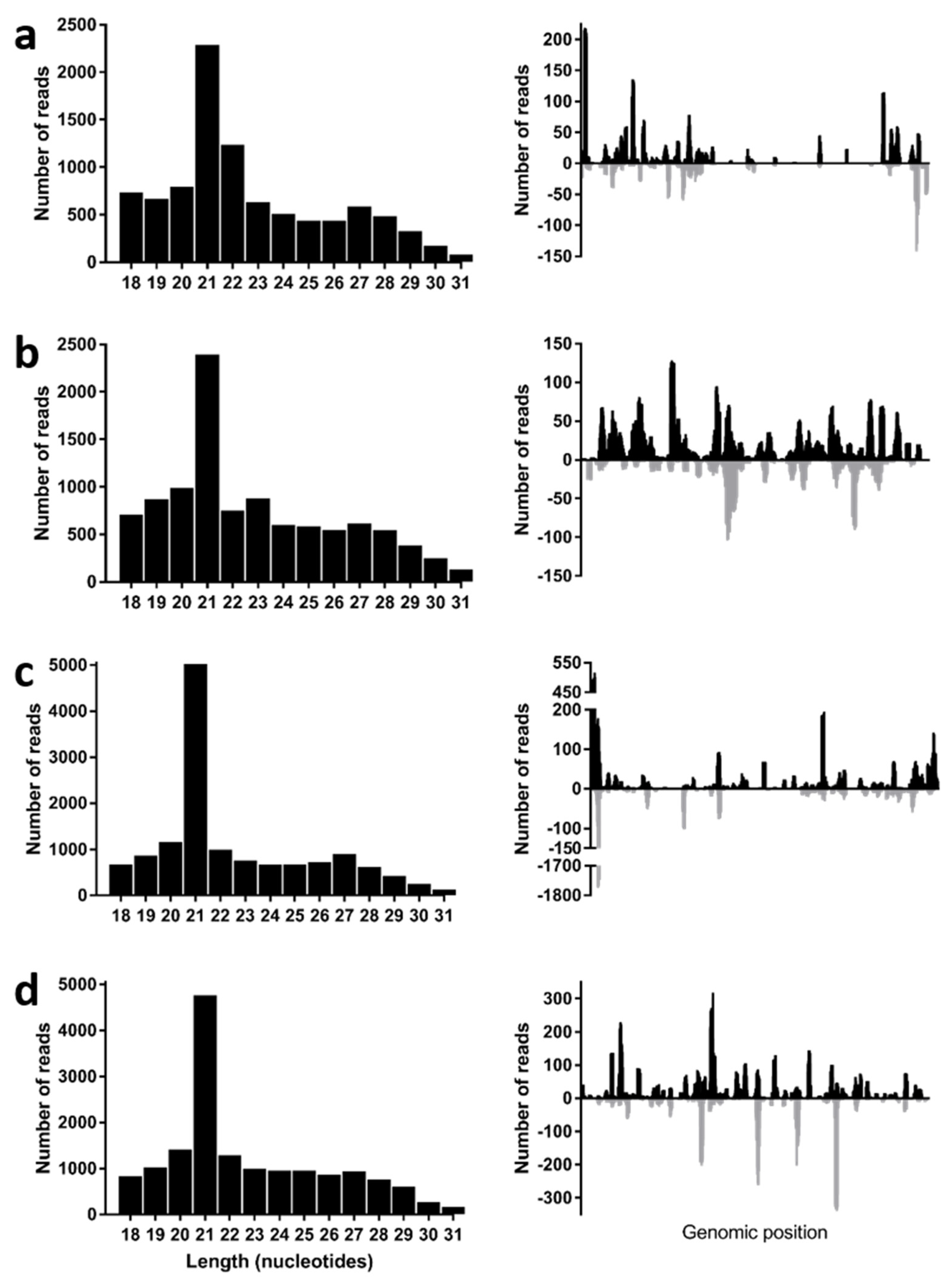
3.4. Virus Derived Small RNA Profiles
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Allsopp, B.A.; Allsopp, M.T. Theileria parva: Genomic DNA studies reveal intra-specific sequence diversity. Mol. Biochem. Parasitol. 1988, 28, 77–83. [Google Scholar] [CrossRef]
- Etebari, K.; Lindsay, K.R.; Ward, A.L.; Furlong, M.J. Australian Sugarcane Soldier Fly’s Salivary Gland Transcriptome in Response to Starvation and Feeding on Sugarcane Crops. Insect Sci. 2020, 27, 708–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hitchcock, B.E. Studies on the life-cycles of two species of soldier flies (diptera, stratiomyidae) which affect sugar-cane in queensland. Bull. Entomol. Res. 1976, 65, 573–578. [Google Scholar] [CrossRef]
- Gordon, K.H.J.; Waterhouse, P.M. Small RNA Viruses of Insects: Expression in Plants and RNA Silencing. Adv. Virus Res. 2006, 68, 459–502. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.-C.; Shi, M.; Tian, J.-H.; Lin, X.-D.; Gao, D.-Y.; He, J.-R.; Wang, J.-B.; Li, C.-X.; Kang, Y.-J.; Yu, B.; et al. A Tick-Borne Segmented RNA Virus Contains Genome Segments Derived from Unsegmented Viral Ancestors. Proc. Natl. Acad. Sci. USA 2014, 111, 6744–6749. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.-D.; Wang, B.; Wei, F.; Han, S.-Z.; Zhang, L.; Yang, Z.-T.; Yan, Y.; Lv, X.-L.; Li, L.; Wang, S.-C.; et al. A New Segmented Virus Associated with Human Febrile Illness in China. N. Engl. J. Med. 2019, 380, 2116–2125. [Google Scholar] [CrossRef]
- Kholodilov, I.S.; Belova, O.A.; Morozkin, E.S.; Litov, A.G.; Ivannikova, A.Y.; Makenov, M.T.; Shchetinin, A.M.; Aibulatov, S.V.; Bazarova, G.K.; Bell-Sakyi, L.; et al. Geographical and Tick-Dependent Distribution of Flavi-Like Alongshan and Yanggou Tick Viruses in Russia. Viruses 2021, 13, 458. [Google Scholar] [CrossRef]
- Kobayashi, D.; Kuwata, R.; Kimura, T.; Shimoda, H.; Fujita, R.; Faizah, A.N.; Kai, I.; Matsumura, R.; Kuroda, Y.; Watanabe, S.; et al. Detection of Jingmenviruses in Japan with Evidence of Vertical Transmission in Ticks. Viruses 2021, 13, 2547. [Google Scholar] [CrossRef]
- Temmam, S.; Bigot, T.; Chrétien, D.; Gondard, M.; Pérot, P.; Pommelet, V.; Dufour, E.; Petres, S.; Devillers, E.; Hoem, T.; et al. Insights into the Host Range, Genetic Diversity, and Geographical Distribution of Jingmenviruses. mSphere 2019, 4, e00645-19. [Google Scholar] [CrossRef] [Green Version]
- Vandegrift, K.J.; Kumar, A.; Sharma, H.; Murthy, S.; Kramer, L.D.; Ostfeld, R.; Hudson, P.J.; Kapoor, A. Presence of Segmented Flavivirus Infections in North America. Emerg. Infect. Dis. 2020, 26, 1810–1817. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, N.; Wang, Z.; Liu, Q. The Discovery of Segmented Flaviviruses: Implications for Viral Emergence. Curr. Opin. Virol. 2020, 40, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, S.R.; Castro-Jorge, L.A.; Ribeiro, J.M.C.; Gardinassi, L.G.; Garcia, G.R.; Brandão, L.G.; Rodrigues, A.R.; Okada, M.I.; Abrão, E.P.; Ferreira, B.R.; et al. Characterisation of Divergent Flavivirus NS3 and NS5 Protein Sequences Detected in Rhipicephalus Microplus Ticks from Brazil. Mem. Inst. Oswaldo Cruz 2014, 109, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Marciel de Souza, W.; Fumagalli, M.J.; Carrasco, A.D.O.T.; Romeiro, M.F.; Modha, S.; Seki, M.C.; Gheller, J.M.; Daffre, S.; Nunes, M.R.T.; Murcia, P.R.; et al. Viral Diversity of Rhipicephalus Microplus Parasitizing Cattle in Southern Brazil. Sci. Rep. 2018, 8, 16315. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Ding, M.; Tan, Z.; Zhao, Z.; Xu, L.; Wu, J.; He, B.; Tu, C. Virome Analysis of Tick-Borne Viruses in Heilongjiang Province, China. Ticks Tick-Borne Dis. 2019, 10, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Dinçer, E.; Hacıoğlu, S.; Kar, S.; Emanet, N.; Brinkmann, A.; Nitsche, A.; Özkul, A.; Linton, Y.-M.; Ergünay, K. Survey and Characterization of Jingmen Tick Virus Variants. Viruses 2019, 11, 1071. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Shen, S.; Wu, H.; Zhang, Y.; Deng, F. Metagenomic Profiling of Viruses Associated with Rhipicephalus Microplus Ticks in Yunnan Province, China. Virol. Sin. 2021, 36, 623–635. [Google Scholar] [CrossRef]
- Ladner, J.T.; Wiley, M.R.; Beitzel, B.; Auguste, A.J.; Dupuis, A.P.; Lindquist, M.E.; Sibley, S.D.; Kota, K.P.; Fetterer, D.; Eastwood, G.; et al. A Multicomponent Animal Virus Isolated from Mosquitoes. Cell Host Microbe 2016, 20, 357–367. [Google Scholar] [CrossRef] [Green Version]
- Teufel, F.; Almagro Armenteros, J.J.; Johansen, A.R.; Gíslason, M.H.; Pihl, S.I.; Tsirigos, K.D.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 6.0 Predicts All Five Types of Signal Peptides Using Protein Language Models. Nat. Biotechnol. 2022. [Google Scholar] [CrossRef]
- Amoa-Bosompem, M.; Kobayashi, D.; Murota, K.; Faizah, A.N.; Itokawa, K.; Fujita, R.; Osei, J.H.N.; Agbosu, E.; Pratt, D.; Kimura, S.; et al. Entomological Assessment of the Status and Risk of Mosquito-Borne Arboviral Transmission in Ghana. Viruses 2020, 12, 147. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, E.; Caudy, A.A.; Hammond, S.M.; Hannon, G.J. Role for a Bidentate Ribonuclease in the Initiation Step of RNA Interference. Nature 2001, 409, 363–366. [Google Scholar] [CrossRef]
- Brackney, D.E.; Beane, J.E.; Ebel, G.D. RNAi Targeting of West Nile Virus in Mosquito Midguts Promotes Virus Diversification. PLoS Pathog. 2009, 5, e1000502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Mierlo, J.T.; van Cleef, K.W.R.; van Rij, R.P. Defense and Counterdefense in the RNAi-Based Antiviral Immune System in Insects. Methods Mol. Biol. Clifton NJ 2011, 721, 3–22. [Google Scholar] [CrossRef]
- Colmant, A.M.G.; Hobson-Peters, J.; Bielefeldt-Ohmann, H.; van den Hurk, A.F.; Hall-Mendelin, S.; Chow, W.K.; Johansen, C.A.; Fros, J.; Simmonds, P.; Watterson, D.; et al. A New Clade of Insect-Specific Flaviviruses from Australian Anopheles Mosquitoes Displays Species-Specific Host Restriction. mSphere 2017, 2, e00262-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colmant, A.M.G.; Etebari, K.; Webb, C.E.; Ritchie, S.A.; Jansen, C.C.; van den Hurk, A.F.; Bielefeldt-Ohmann, H.; Hobson-Peters, J.; Asgari, S.; Hall, R.A. Discovery of New Orbiviruses and Totivirus from Anopheles Mosquitoes in Eastern Australia. Arch. Virol. 2017, 162, 3529–3534. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.-D.; Vasilakis, N.; Tian, J.-H.; Li, C.-X.; Chen, L.-J.; Eastwood, G.; Diao, X.-N.; Chen, M.-H.; Chen, X.; et al. Divergent Viruses Discovered in Arthropods and Vertebrates Revise the Evolutionary History of the Flaviviridae and Related Viruses. J. Virol. 2016, 90, 659–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaafar, Y.Z.A.; Ziebell, H. Comparative Study on Three Viral Enrichment Approaches Based on RNA Extraction for Plant Virus/Viroid Detection Using High-Throughput Sequencing. PLoS ONE 2020, 15, e0237951. [Google Scholar] [CrossRef] [PubMed]
- Ishiwata, K.; Sasaki, G.; Ogawa, J.; Miyata, T.; Su, Z.-H. Phylogenetic Relationships among Insect Orders Based on Three Nuclear Protein-Coding Gene Sequences. Mol. Phylogenet. Evol. 2011, 58, 169–180. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.-D.; Tian, J.-H.; Chen, L.-J.; Chen, X.; Li, C.-X.; Qin, X.-C.; Li, J.; Cao, J.-P.; Eden, J.-S.; et al. Redefining the Invertebrate RNA Virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef]
- Paraskevopoulou, S.; Käfer, S.; Zirkel, F.; Donath, A.; Petersen, M.; Liu, S.; Zhou, X.; Drosten, C.; Misof, B.; Junglen, S. Viromics of Extant Insect Orders Unveil the Evolution of the Flavi-like Superfamily. Virus Evol. 2021, 7, veab030. [Google Scholar] [CrossRef]
- Rodriguez-Romero, J.; Chiapello, M.; Cordoba, L.; Turina, M.; Ayllon, M.A. The Miscellaneous Mycovirome Associated to the Plant Pathogenic Fungus Erysiphe Necator. 2021. Available online: http://www.ncbi.nlm.nih.gov/nuccore/MN558700.1 (accessed on 15 November 2021).
- Chiapello, M.; Rodríguez-Romero, J.; Ayllón, M.A.; Turina, M. Analysis of the Virome Associated to Grapevine Downy Mildew Lesions Reveals New Mycovirus Lineages. Virus Evol. 2020, 6, veaa058. [Google Scholar] [CrossRef]
- Brackney, D.E.; Scott, J.C.; Sagawa, F.; Woodward, J.E.; Miller, N.A.; Schilkey, F.D.; Mudge, J.; Wilusz, J.; Olson, K.E.; Blair, C.D.; et al. C6/36 Aedes Albopictus Cells Have a Dysfunctional Antiviral RNA Interference Response. PLoS Negl. Trop. Dis. 2010, 4, e856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.S.; Nakahara, K.; Pham, J.W.; Kim, K.; He, Z.; Sontheimer, E.J.; Carthew, R.W. Distinct Roles for Drosophila Dicer-1 and Dicer-2 in the SiRNA/MiRNA Silencing Pathways. Cell 2004, 117, 69–81. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Segment | ORF | Direction | Name | Sequence | Product Size |
|---|---|---|---|---|---|
| 1 | NSP1 | Forward | IFJV1 seg1 1F | CCTGGAAGATGTATGGTGTGATTGG | 514 bp |
| Reverse | IFJV1 seg1 1R | GCTCCTTTCCCTGTTCTATCTTGG | |||
| 3 | NSP2 | Forward | IFJV1 seg3 1F | GGAAGACTCAAACAGAATCTCATGC | 542 bp |
| Reverse | IFJV1 seg3 1R | GGTACTTCGCATGTCACATGC | |||
| 4 | VP2 | Forward | IFJV1 seg4 1F | GGTAGCAAGTTACAAGATGG | 523 bp |
| Reverse | IFJV1 seg4 1R | CATACACAACATCTCCATATGTGTGG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colmant, A.M.G.; Furlong, M.J.; Etebari, K. Discovery of a Novel Jingmenvirus in Australian Sugarcane Soldier Fly (Inopus flavus) Larvae. Viruses 2022, 14, 1140. https://doi.org/10.3390/v14061140
Colmant AMG, Furlong MJ, Etebari K. Discovery of a Novel Jingmenvirus in Australian Sugarcane Soldier Fly (Inopus flavus) Larvae. Viruses. 2022; 14(6):1140. https://doi.org/10.3390/v14061140
Chicago/Turabian StyleColmant, Agathe M. G., Michael J. Furlong, and Kayvan Etebari. 2022. "Discovery of a Novel Jingmenvirus in Australian Sugarcane Soldier Fly (Inopus flavus) Larvae" Viruses 14, no. 6: 1140. https://doi.org/10.3390/v14061140
APA StyleColmant, A. M. G., Furlong, M. J., & Etebari, K. (2022). Discovery of a Novel Jingmenvirus in Australian Sugarcane Soldier Fly (Inopus flavus) Larvae. Viruses, 14(6), 1140. https://doi.org/10.3390/v14061140