
Infectious Spleen and Kidney Necrosis Virus (ISKNV) Triggers Mitochondria-Mediated Dynamic Interaction Signals via an Imbalance of Bax/Bak over Bcl-2/Bcl-xL in Fish Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals, Drugs and Antibodies Used
2.2. Cell Culture and Virus
2.3. Cell Viability
2.4. Annexin V-FLUOS Staining
2.5. Evaluation of Mitochondrial Membrane Potential Using a Lipophilic Cationic Dye
2.6. Western Blotting Analysis
2.7. Statistical Analyses
3. Results
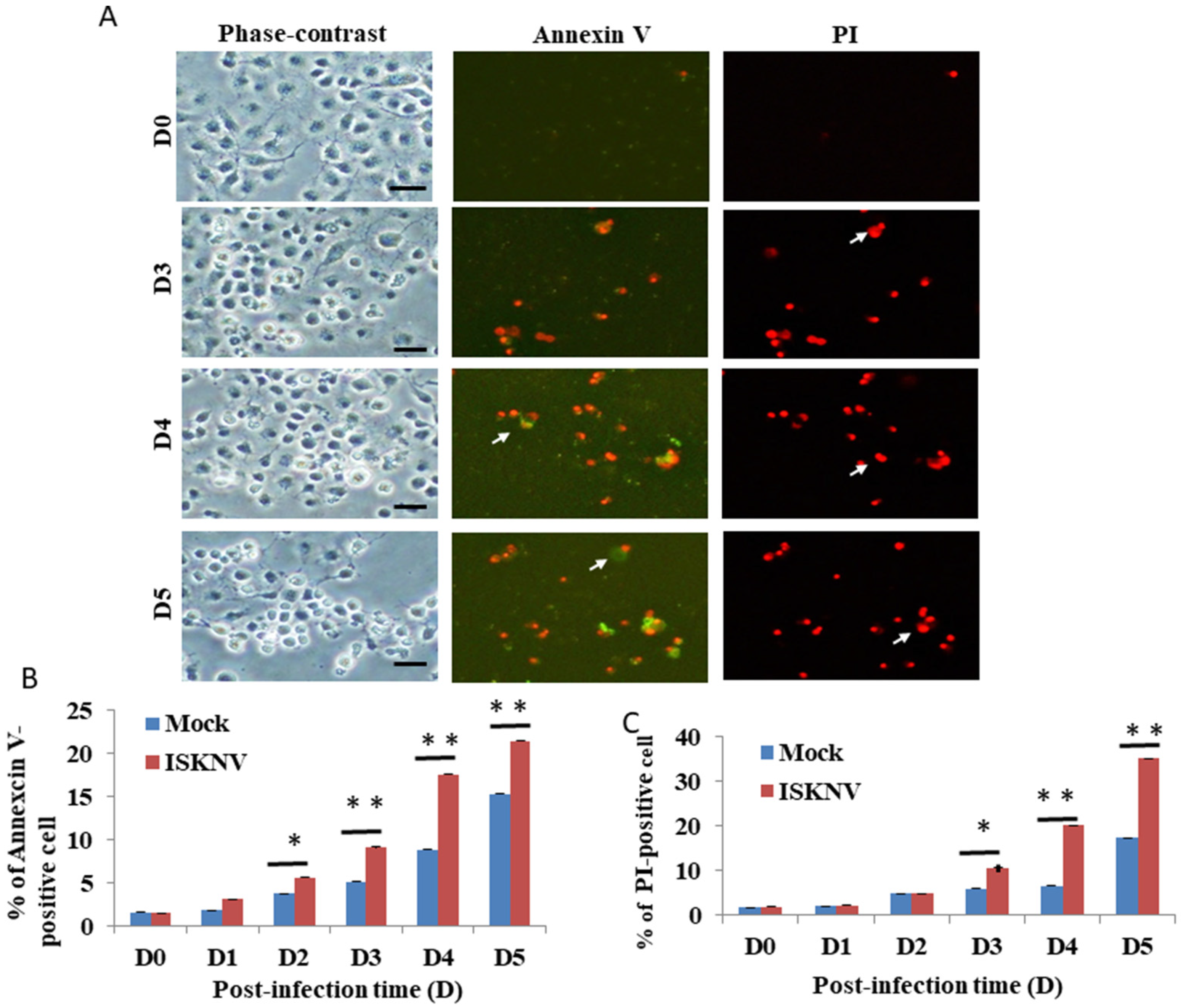
3.1. ISKNV Induces Host Apoptotic Cell Death in GF-1 Cells
3.2. ISKNV Can Induce Apoptotic Cell Death and Necrosis in GF-1 Cells
3.3. ISKNV Induces Mitochondrial MMP Loss
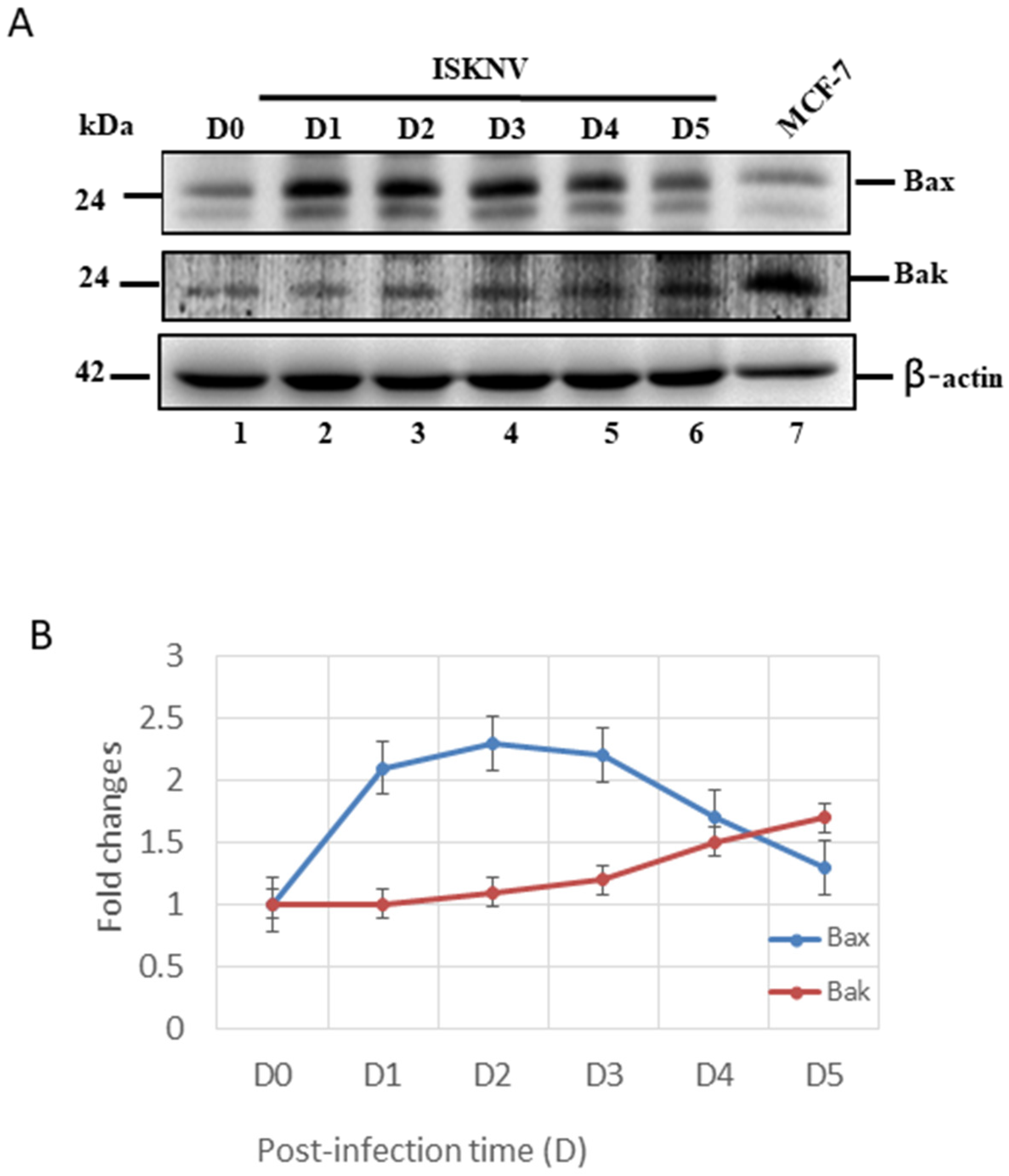
3.4. ISKNV Infection Upregulates Both Anti-Apoptotic Bcl-2-Family Members’ (Bcl-2/Bcl-xL) and Pro-Apoptotic Members’ (Bax/Bak) Expression
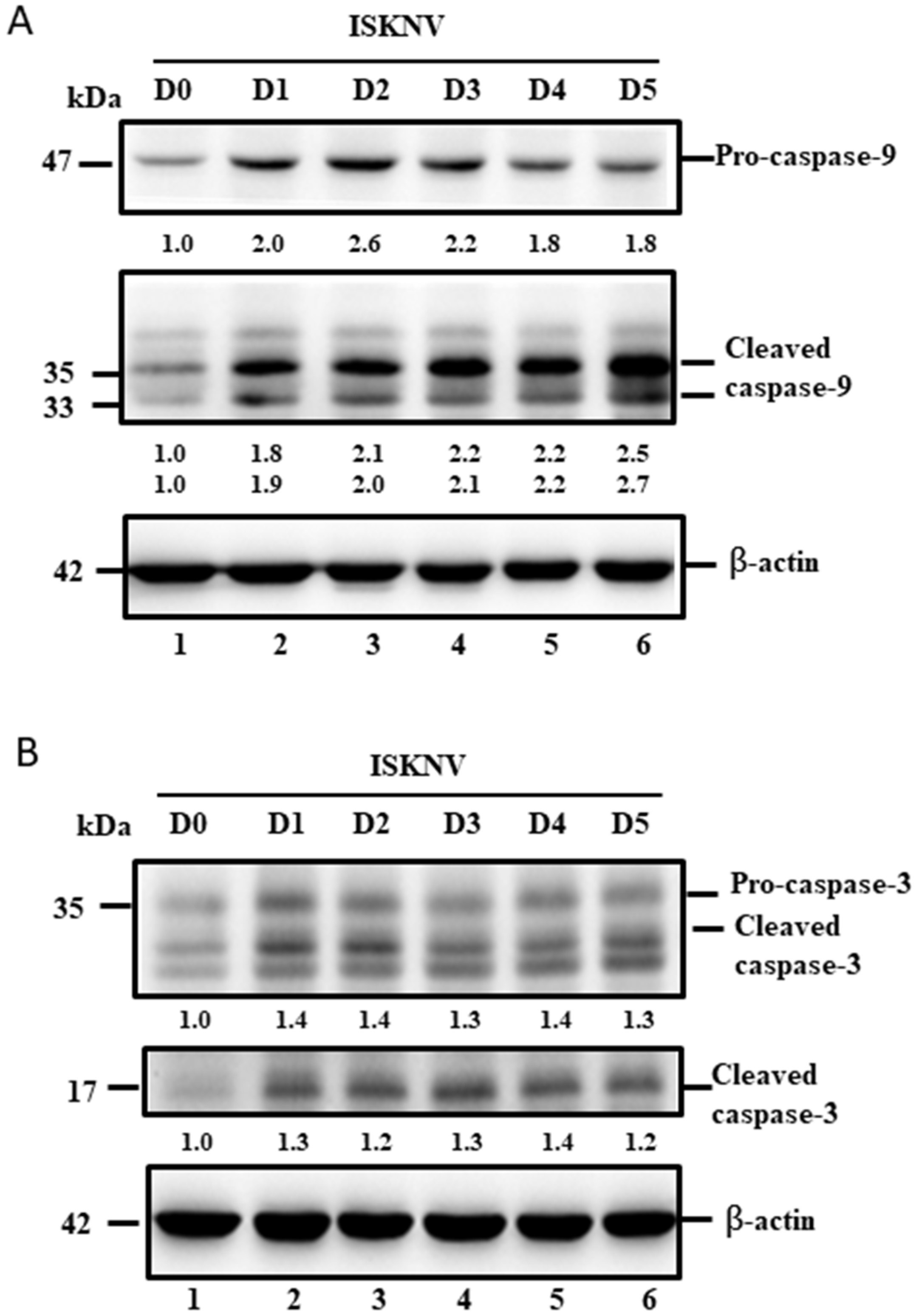
3.5. ISKNV Induces Caspase-9 and Caspase-3 Activation at the Replication Stage
4. Discussion
4.1. ISKNV Induced Annexin-V/PI Positives in GF-1 Cells
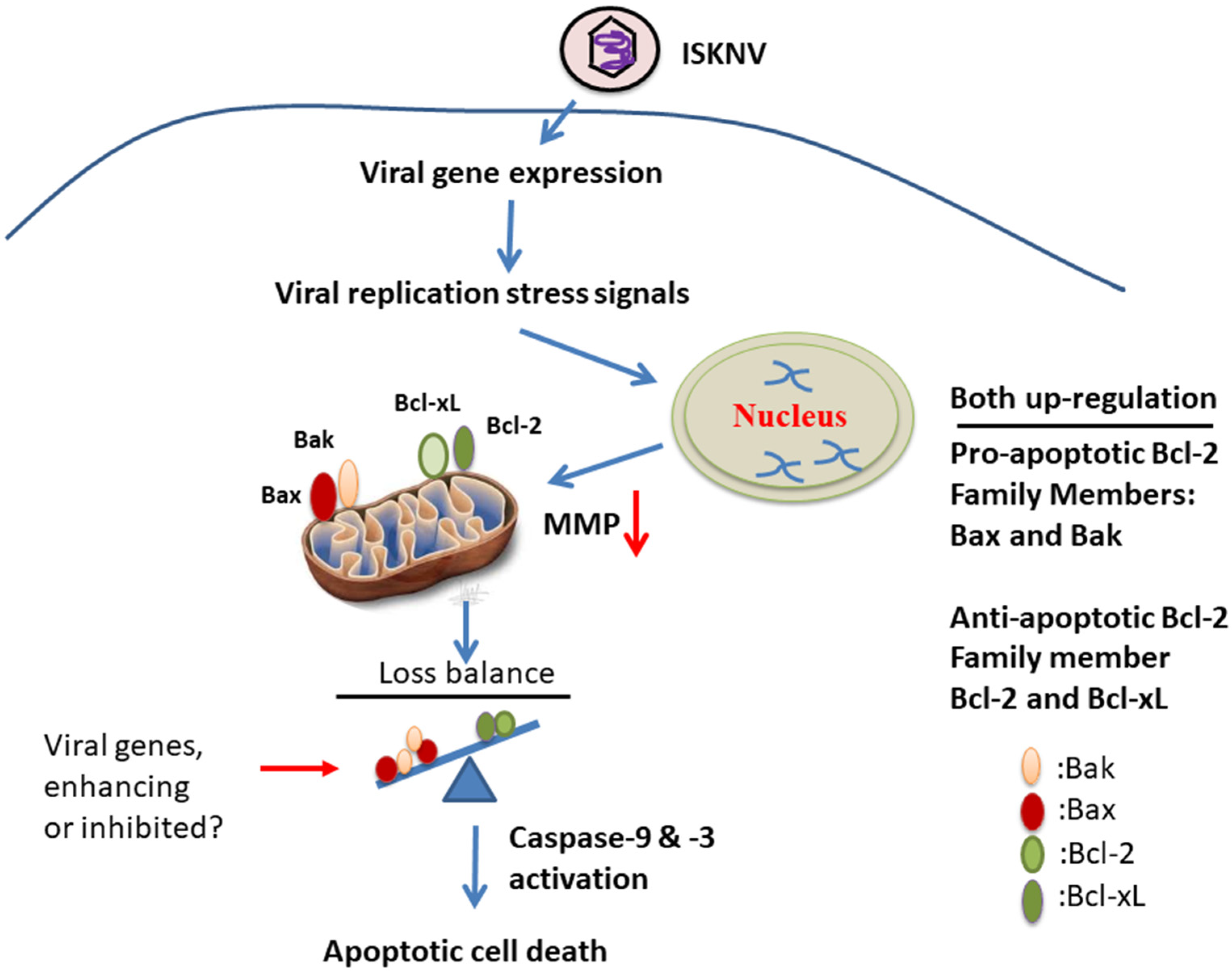
4.2. What Kind of Cell Death Signaling Interaction Occurs during ISKNV-Triggered MMP Loss?
4.3. Are Bcl-2-Family Members Important in ISKN-Induced Cell Death?
4.4. Does the Viral Genome Provide Cell Death or Anti-Cell Death Signals during the Viral Replication Cycle?
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kurita, J.; Nakajima, K. Review: Megalocytivirus. Viruses 2012, 4, 521–538. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, K.; Shariff, M.; Omar, A.R.; Hair-Bejo, M. Megalocytivirus infection in fish. Rev. Aquac. 2012, 4, 221–233. [Google Scholar] [CrossRef]
- Williams, T.; Barbosa, S.V.; Chinchar, V.G. A decade of advances in iridovirus research. Adv. Virus Res. 2005, 65, 173–248. [Google Scholar] [PubMed]
- Huang, X.H.; Huang, Y.H.; Ouyang, Z.L.; Xu, L.X.; Yan, Y.; Cui, H.C.; Han, X.; Qin, Q.W. Singapore grouper iridovirus, a large DNA virus, induces non apoptotic cell death by a cell type dependent fashion and evokes ERK signaling. Apoptosis 2011, 16, 831–845. [Google Scholar] [CrossRef]
- Pham, P.H.; Lai, Y.S.; Lee, F.F.; Bols, N.C.; Chiou, P.P. Differential viral propagation and induction of apoptosis by grouper iridovirus (GIV) in cell lines from three non-host species. Virus Res. 2012, 167, 16–25. [Google Scholar] [CrossRef]
- Kumar, S. Caspase function in programmed cell death. Cell Death Differ. 2006, 14, 32–43. [Google Scholar] [CrossRef]
- Ameisen, J.C. On the origin, evolution, and nature of programmed cell death: A timeline of four billion years. Cell Death Differ. 2002, 9, 367–393. [Google Scholar] [CrossRef]
- Clarke, P.; Tyler, K.L. Apoptosis in animal models of virus induced disease. Nat. Rev. Microbiol. 2009, 7, 144–155. [Google Scholar] [CrossRef]
- Wyllie, A.H.; Kerr, J.F.R.; Currie, A.R. Cell death: The significance of apoptosis. Int. Rev. Cytol. 1980, 68, 251–306. [Google Scholar]
- White, E. Life, death, and the pursuit of apoptosis. Genes Dev. 1996, 10, 1–15. [Google Scholar] [CrossRef]
- Chen, P.C.; Wu, J.L.; Her, G.M.; Hong, J.R. Aquatic birnavirus induces necrotic cell death via mitochondria-mediated caspases pathway that is inhibited by bongkrekic acid. Fish Shellfish Immunol. 2010, 28, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Ferri, K.F.; Kroemer, G. Organelle-specific initiation of cell death pathways. Nat. Cell Biol. 2001, 3, E255–E263. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Giorgi, C.; Pinton, P. Molecular mechanisms and consequences of mitochondrial permeability transition. Nat. Rev. Mol. Cell Biol. 2022, 23, 266–285. [Google Scholar] [CrossRef]
- Hong, J.R. Betanodavirus: Mitochondrial disruption and necrotic cell death. World J. Virol. 2013, 2, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.C.; Gong, H.Y.; Cheng, C.Y.; Wang, J.P.; Hong, J.R.; Wu, J.L. Cloning and characterization of zfBLP1, zfBcl-xL homologue from the zebrafish. Danio Rerio. Biochim. Biophys. Acta 2001, 1519, 127–133. [Google Scholar] [CrossRef]
- Zamzami, N.; Kroemer, G. The mitochondrion in apoptosis: How Pandora’s box opens. Nat. Rev. Mol. Cell Biol. 2001, 2, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.R.; Wu, J.L. Induction of apoptotic death in cells via bad gene expression by infectious pancreatic necrosis virus infection. Cell Death Differ. 2002, 9, 113–124. [Google Scholar] [CrossRef]
- Chen, X.Y.; Wen, C.M.; Wu, J.L.; Su, Y.C.; Hong, J.R. Giant seaperch iridovirus infection upregulates Bax and Bak expression, leading to apoptotic death of fish cells. Fish Shellfish Immunol. 2015, 45, 848–857. [Google Scholar] [CrossRef]
- Chen, X.Y.; Wen, C.W.; Wu, J.L.; Su, Y.C.; Hong, J.R. Giant seaperch iridovirus induces mitochondria-mediated cell death that is suppressed by bongkrekic acid and cycloheximide in fish cell line. Virus Res. 2016, 213, 37–75. [Google Scholar] [CrossRef]
- Shiu, J.Y.; Hong, J.R.; Ku, C.C.; Wen, C.M. Complete genome sequence and phylogenetic analysis of megalocytivirus RSIV-Ku: A natural recombination infectious spleen and kidney necrosis virus. Arch. Virol. 2018, 163, 1037–1042. [Google Scholar] [CrossRef]
- Labbe, K.; Saleh, M. Cell death in the host response to infection. Cell Death Differ. 2008, 15, 1339–1349. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.P.; Wu, J.L.; Su, Y.C.; Hong, J.R. Anti-Bcl-2 family members, zfBcl-xL and zfMcl-1a, prevent cytochrome c release from cells undergoing betanodavirus induced secondary necrotic cell death. Apoptosis 2007, 12, 1043–1060. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.R.; Huang, L.J.; Wu, J.L. Fish birnavirus induces apoptotic through activated caspases 8 and 3 in zebrafish cell line. J. Fish Dis. 2005, 28, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.R.; Gong, H.Y.; Wu, J.L. VP5, a novel anti-apoptosis of Bcl-2 family member that up-regulates the Mcl-1 and limit-regulates the viral proteins expression. Virology 2002, 295, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.W.; Chiu, H.C.; Chiu, Y.W.; Wu, J.L.; Hong, J.R. EPA and DHA can enhance host cell survival via modulation of Fas/tBid-mediated death pathway with ISKNV infection in GF-1 cells. Fish Shellfish Immunol. 2020, 9, 608–616. [Google Scholar] [CrossRef]
- Dobos, P.; Hill, B.; Hallett, R.; Kells, D.; Becht, H.; Teninges, D. Biophysica and biochemical characterization of five animal viruses with bi-segmented double stranded RNA genomes. J. Virol. 1979, 32, 593–605. [Google Scholar] [CrossRef]
- Mullen, P.D.; Brand, R.J.; Parlette, G.N. Evaluation of dye exclusion and colony inhibition techniques for detection of polyoma-specific, cell-mediated immunity. J. Natl. Cancer Inst. 1975, 54, 229.e231. [Google Scholar] [CrossRef]
- Hong, J.R.; Lin, T.L.; Yang, J.Y.; Hsu, Y.L.; Wu, J.L. Dynamics of non-typical apoptotic morphological changes visualized by green fluorescent protein in living cells with infectious pancreatic necrosis virus infection. J. Virol. 1999, 73, 5056–5063. [Google Scholar] [CrossRef]
- Chen, S.P.; Yang, H.L.; Her, G.M.; Lin, H.Y.; Jeng, M.F.; Wu, J.L.; Hong, J.R. Betanodavirus induces phosphatidylserine exposure and loss of mitochondrial membrane potential in secondary necrotic cells, both of which are blocked by bongkrekic acid. Virology 2006, 347, 379–391. [Google Scholar] [CrossRef]
- Jancovich, J.K.; Chinchar, V.G.; Hyatt, A.; Miyazaki, T.; Williams, T. Family iridoviridae. In Virus Taxonomy: 9th Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Lefkowitz, E., Adams, M.J., Carstens, E.B., Eds.; Elsevier: San Diego, CA, USA, 2011; pp. 193–210. [Google Scholar]
- Huang, Y.H.; Huang, X.H.; Cai, J.; Ye, F.Z.; Qin, Q.W. Involvement of the mitogen-activated protein kinase pathway in soft-shelled turtle iridovirus-induced apoptosis. Apoptosis 2011, 16, 581–593. [Google Scholar] [CrossRef]
- Lee, A.J.; Liao, H.J.; Hong, J.R. Overexpression of Bcl2 and Bcl2L1 can suppress betanodavirus-induced type III cell death and autophagy induction in GF-1 cells. Symmetry 2022, 14, 360. [Google Scholar] [CrossRef]
- Chitnis, N.S.; D’Costa, S.M.; Paul, E.R.; Bilimoria, S.L. Modulation of iridovirus induced apoptosis by endocytosis, early expression, JNK, and apical caspase. Virology 2008, 370, 333–342. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Huang, X.; Wang, W.; Huang, Y.; Xu, L.; Qin, Q. Involvement of the PI3K and ERK signaling pathways in largemouth bass virus-induced apoptosis and viral replication. Fish Shellfish Immunol. 2014, 41, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.B.; Cong, R.S.; Fan, T.J.; Mei, X.G. Induction of apoptosis in a flounder gill cell line by lymphocystis disease virus infection. J. Fish Dis. 2004, 27, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Chinchar, V.G.; Bryan, L.; Wang, J.; Long, S.; Chinchar, G.D. Induction of apoptosis in frog virus 3-infected cells. Virology 2003, 306, 303–312. [Google Scholar] [CrossRef]
- Gray, M.J.; Miller, D.L.; Hoverman, J.T. Ecology and pathology of amphibian ranaviruses. Dis. Aquat. Organ. 2009, 87, 243–266. [Google Scholar] [CrossRef]
- Whittington, R.J.; Becker, J.A.; Dennis, M.M. Iridovirus infections in finfish ecritical review with emphasis on ranaviruses. J. Fish Dis. 2010, 33, 95–122. [Google Scholar] [CrossRef]
- Benedict, C.A.; Norris, P.S.; Ware, C.F. To kill or be killed: Viral evasion of apoptosis. Nat Immunol. 2002, 3, 1013–1018. [Google Scholar] [CrossRef]
- Vanlandschoot, P.; Leroux-Roels, G. Viral apoptotic mimicry: An immune evasion strategy developed by the hepatitis B virus? Trends Immunol. 2003, 24, 144–147. [Google Scholar] [CrossRef]
- Levine, A.J. P53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef]
- Eisenberg-Bord, M.; Schuldiner, M. Ground control to major TOM: Mitochondria-nucleus communication. FEBS J. 2017, 284, 196.e210. [Google Scholar] [CrossRef] [PubMed]
- Gottschling, D.E.; Nystrom, T. The upsides and downsides of organelle interconnectivity. Cell 2017, 169, 24.e34. [Google Scholar] [CrossRef] [PubMed]
- Boldt, K.; van Reeuwijk, J.; Lu, Q.; Koutroumpas, K.; Nguyen, T.M.; Texier, Y.; van Beersum, S.E.C.; Horn, N.; Willer, J.R.; Mans, D.A.; et al. An organelle-specific protein landscape identifies novel diseases and molecular mechanisms. Nat Commun. 2016, 7, 11491. [Google Scholar] [CrossRef] [PubMed]
- Freed, E.O. HIV-1 and the host cell: An intimate association. Trends Microbiol. 2004, 12, 170.e7. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.A. Virulence and pathogenesis. Trends Microbiol. 2002, 10, 314.e7. [Google Scholar] [CrossRef]
- Mercer, J.; Schelhaas, M.; Helenius, A. Virus entry by endocytosis. Annu. Rev. Biochem. 2010, 79, 803.e33. [Google Scholar] [CrossRef] [PubMed]
- Kagan, J.C. Signaling organelles of the innate immune system. Cell 2012, 151, 1168.e78. [Google Scholar] [CrossRef]
- Tait, S.W.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef]
- Riedl, S.J.; Salvesen, G.S. The apoptosome: Signalling platform of cell death. Nat. Rev. Mol. Cell Biol. 2007, 8, 405–413. [Google Scholar] [CrossRef]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef]
- Shamas-Din, A.; Brahmbhatt, H.; Leber, B.; Andrews, D.W. BH3-only proteins: Orchestrators of apoptosis. Biochim. Biophys. Acta 2011, 1813, 508–520. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Fan, Y.; Zhou, Y.; Jiang, N.; Xue, M.; Meng, Y.; Liu, W.; Zhang, J.; Lin, G.; Lingbing Zeng, L. Bcl-xL Reduces Chinese Giant Salamander Iridovirus-Induced Mitochondrial Apoptosis by Interacting with Bak and Inhibiting the p53 Pathway. Viruses 2021, 13, 2224. [Google Scholar] [CrossRef] [PubMed]
- Wen, C.M.; Hong, J.R. Complete Genome Sequence of a Giant Sea Perch Iridovirus in Kaohsiung, Taiwan. Genome Announc. 2016, 4, e01759-15. [Google Scholar] [CrossRef] [PubMed]
- Banjara, S.; Mao, J.; Ryan, T.M.; Caria, S.; Kvansakul, M. Grouper iridovirus GIV66 is a Bcl-2 protein that inhibits apoptosis by exclusively sequestering Bim. J. Biol. Chem. 2018, 293, 5464–5477. [Google Scholar] [CrossRef]
- Reshi, L.; Wang, H.V.; Hui, C.F.; Su, Y.C.; Hong, H.R. Anti-apoptotic genes Bcl-2 and Bcl-xL overexpression can block iridovirus serine/threonine kinase-induced Bax/mitochondria-mediated cell death in GF-1 cells. Fish Shellfish Immunol. 2017, 61, 120–129. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, P.-H.; Hsueh, T.-C.; Wu, J.-L.; Hong, J.-R. Infectious Spleen and Kidney Necrosis Virus (ISKNV) Triggers Mitochondria-Mediated Dynamic Interaction Signals via an Imbalance of Bax/Bak over Bcl-2/Bcl-xL in Fish Cells. Viruses 2022, 14, 922. https://doi.org/10.3390/v14050922
Chen P-H, Hsueh T-C, Wu J-L, Hong J-R. Infectious Spleen and Kidney Necrosis Virus (ISKNV) Triggers Mitochondria-Mediated Dynamic Interaction Signals via an Imbalance of Bax/Bak over Bcl-2/Bcl-xL in Fish Cells. Viruses. 2022; 14(5):922. https://doi.org/10.3390/v14050922
Chicago/Turabian StyleChen, Pin-Han, Tsai-Ching Hsueh, Jen-Leih Wu, and Jiann-Ruey Hong. 2022. "Infectious Spleen and Kidney Necrosis Virus (ISKNV) Triggers Mitochondria-Mediated Dynamic Interaction Signals via an Imbalance of Bax/Bak over Bcl-2/Bcl-xL in Fish Cells" Viruses 14, no. 5: 922. https://doi.org/10.3390/v14050922
APA StyleChen, P.-H., Hsueh, T.-C., Wu, J.-L., & Hong, J.-R. (2022). Infectious Spleen and Kidney Necrosis Virus (ISKNV) Triggers Mitochondria-Mediated Dynamic Interaction Signals via an Imbalance of Bax/Bak over Bcl-2/Bcl-xL in Fish Cells. Viruses, 14(5), 922. https://doi.org/10.3390/v14050922