
NF-κB-Induced R-Loops and Genomic Instability in HTLV-1-Infected and Adult T-Cell Leukemia Cells

{kind=link}
{kind=link}
Abstract
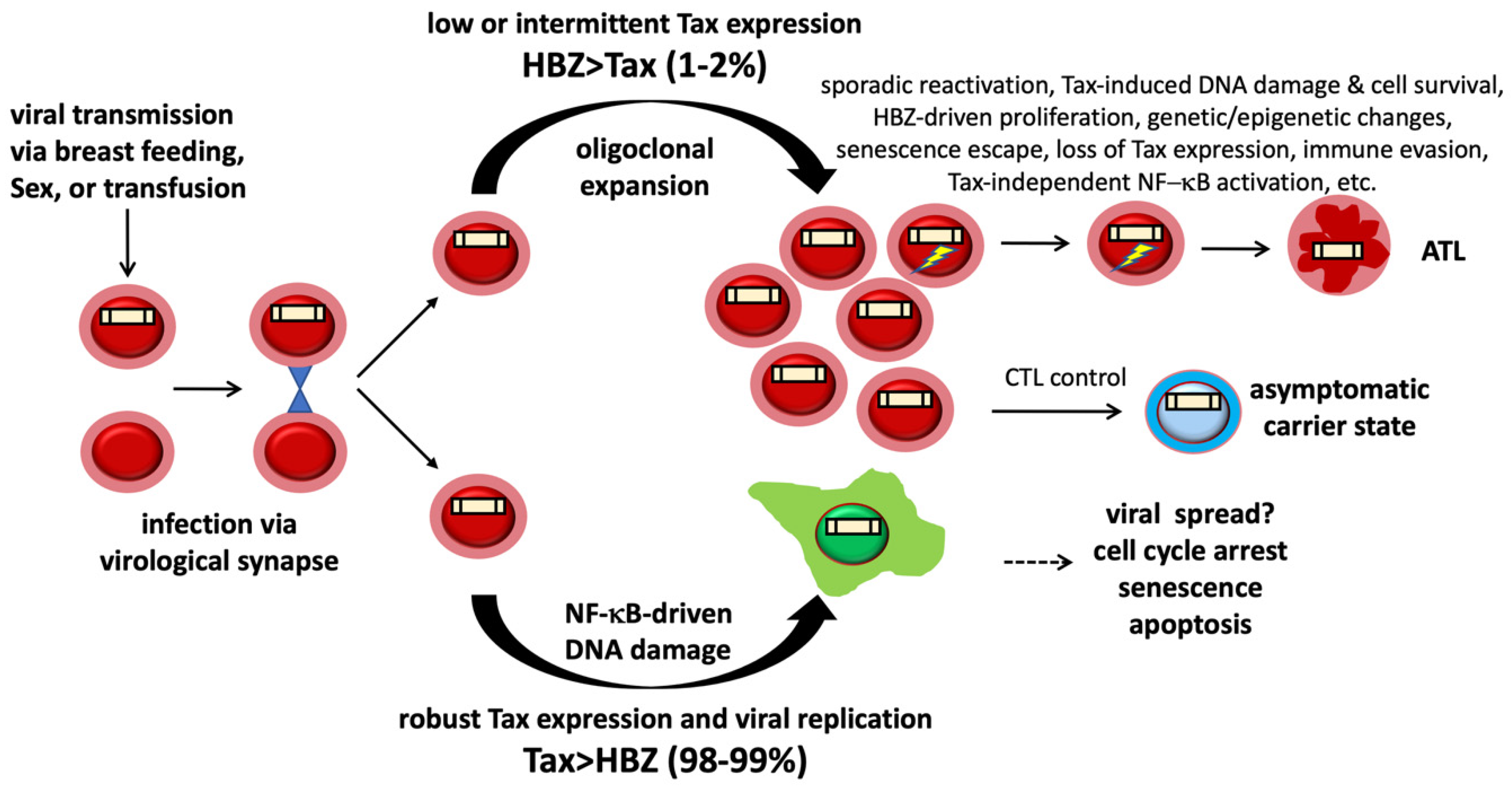
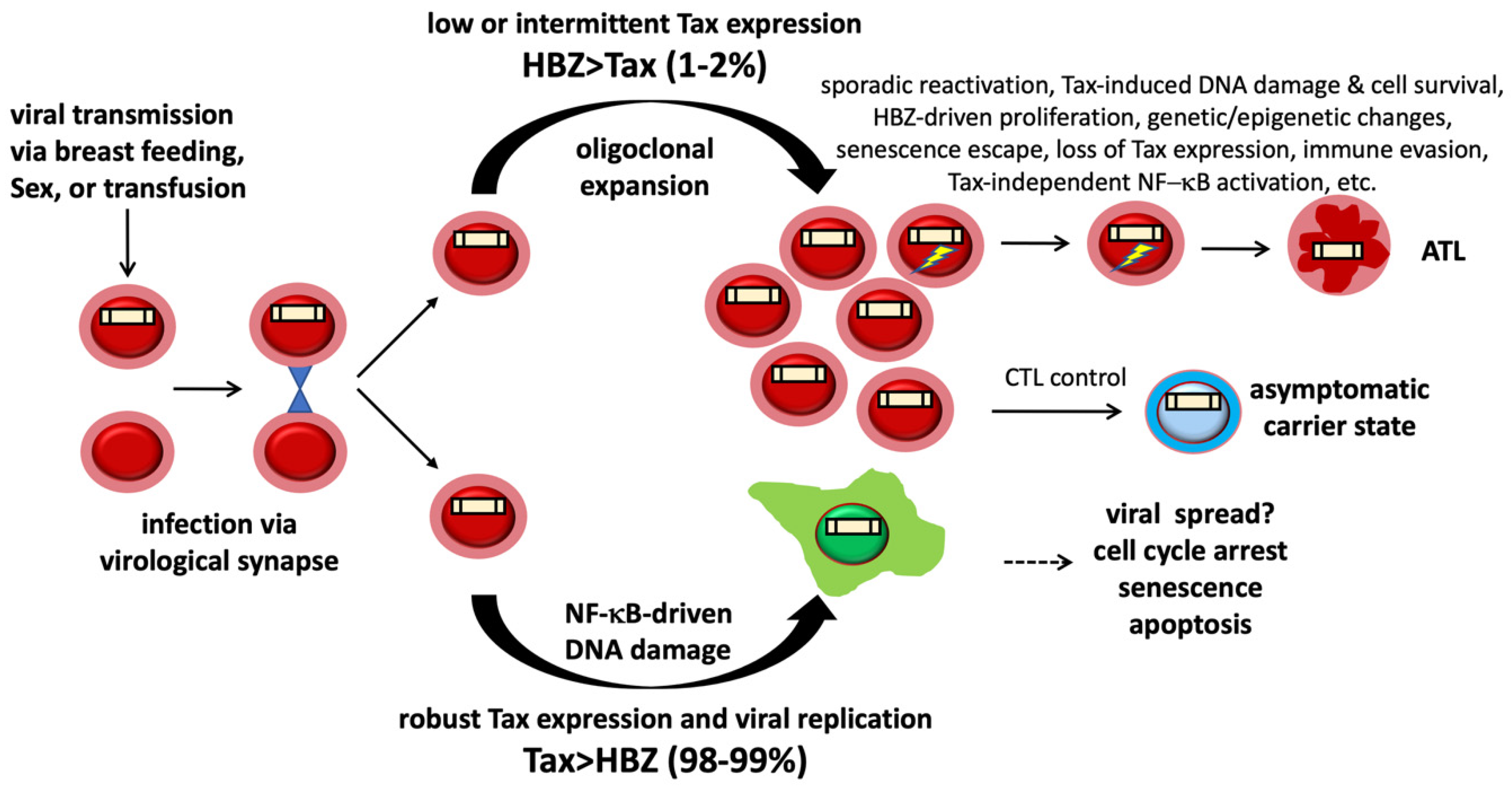
:1. Introduction
2. R-Loops, DNA Double-Strand Breaks, and Genomic Instability
3. Tax Hijacks RNF8 for Canonical NF-κB Activation and DNA Damage Response Perturbation
4. NF-κB Hyperactivation by Tax Induces Cellular Senescence
5. Transcription-Coupled Nucleotide Excision Repair and Transcription-Replication Conflict May Underlie the R-Loop-Associated DNA Double-Strand Breaks Caused by Tax
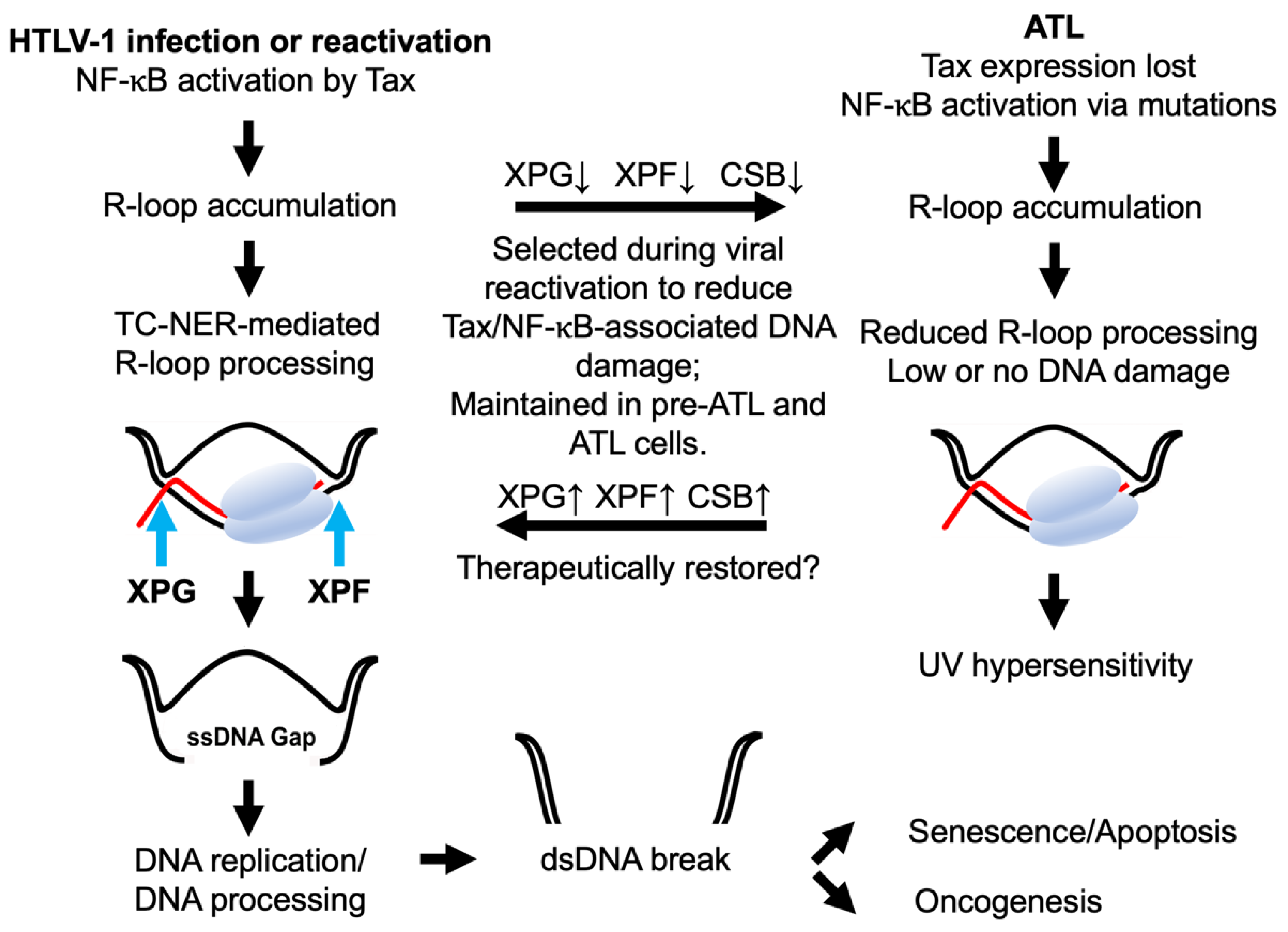
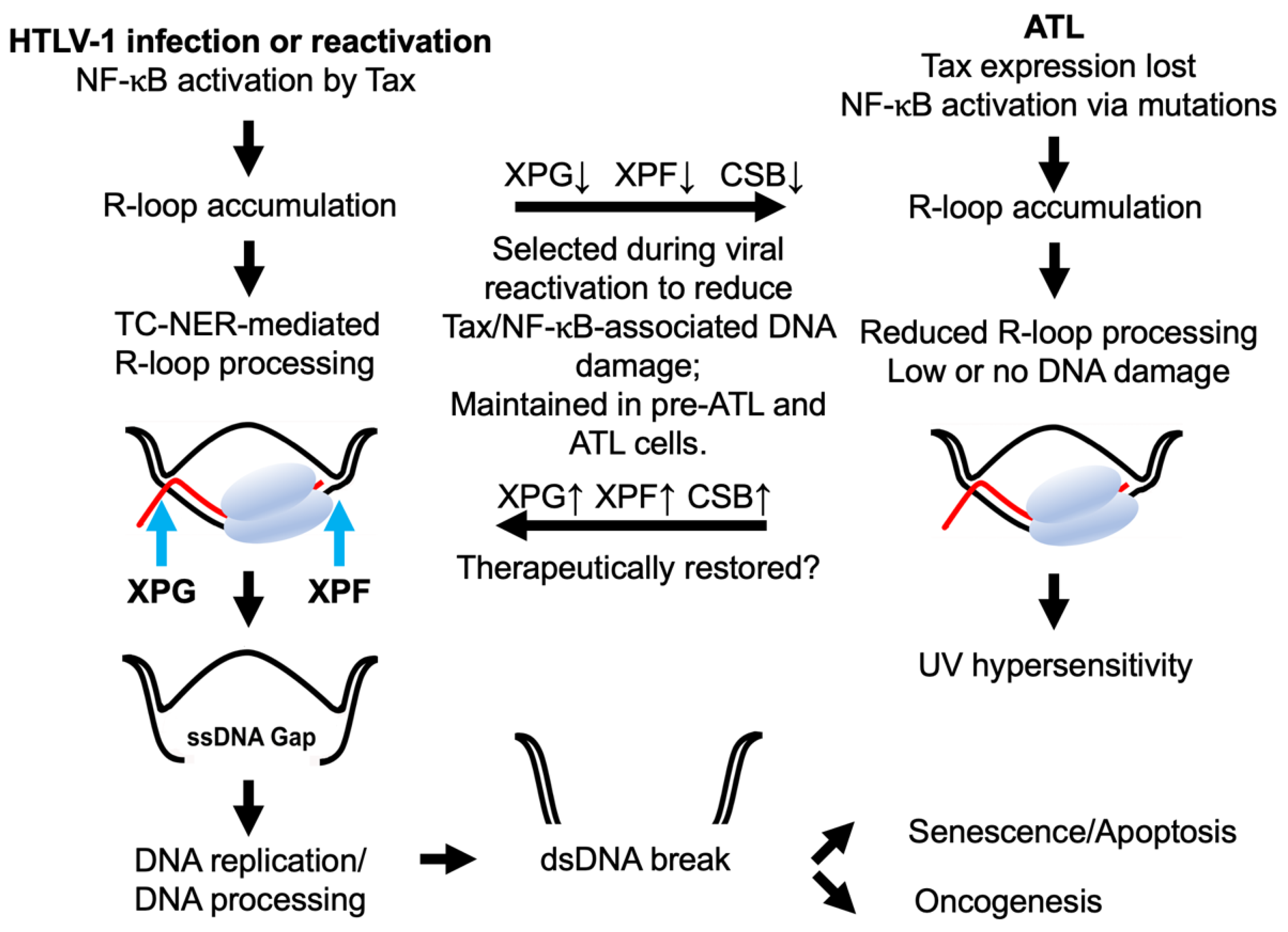
6. NF-κB Hyperactivation and Excess R-Loop Accumulation in ATL Cells
7. NF-κB-Induced R-Loops and DNA Double-Strand Breaks in ATL
8. Concluding Remarks and Future Direction
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Glossary
| ATF-1 | activating transcription factor 1 |
| ATL | adult T cell leukemia |
| B2M | β2 microglobulin |
| BRCA1 | breast cancer gene 1 |
| BRCA2 | breast cancer gene 2 |
| CBP | CREB-binding protein |
| CREB | cAMP response element-binding protein |
| CSB | cockayne syndrome B |
| CTL | cytotoxic T lymphocyte |
| DDR | DNA damage response |
| DHX9 | DEAH-box helicase 9 |
| DNA DSB | DNA double-strand break |
| DNA-PK | DNA-dependent protein kinase |
| FACT complex | facilitates chromatin transactions complex |
| FAS | FS-7-associated surface antigen, a cell surface death receptor |
| GATA3 | GATA binding protein 3 |
| HBZ | HTLV-1 basic domain leucine zipper protein |
| HLA-B | human leukocyte antigen (HLA) complex-B, major histocompatibility complex class I-B |
| HRAS | Harvey rat sarcoma oncogene |
| HTLV-1 | Human T-cell leukemia virus type 1 |
| Indel | Insertion and deletion |
| IKK | Inhibitor of NF-κB kinase |
| JNK | c-Jun N-terminal kinase |
| LUBAC | linear (M1) ubiquitin assembly complex |
| MDC1 | mediator of DNA damage checkpoint 1 |
| mTOR | mammalian target of rapamycin |
| NHEJ | non-homologous end joining |
| NIK | NF-κB-inducing kinase, mitogen-activated protein kinase kinase kinase 14 |
| NF-κB | nuclear factor kappa B |
| NFKBIA | NF-κB Inhibitor α |
| PVL | proviral load |
| RNAPII | RNA polymerase II |
| RNF8 | RING finger protein 8 |
| Sen1 | yeast homolog of senataxin |
| SETX | senataxin, DNA/RNA helicase that resolves R-loops |
| TAK1 | TGFβ-activated kinase 1, mitogen-activated protein kinase kinase kinase 7 |
| Tax | Trans-activator of the X region |
| Tax-IRS | Tax-induced rapid senescence |
| TC-NER | transcription-coupled nucleotide excision repair |
| TNFAIP3 | TNFα-induced protein 3 |
| TP53 | tumor protein p53 (or p53) |
| XPF | xeroderma pigmentosum F |
| XPG | xeroderma pigmentosum G |
References
- Tsukasaki, K.; Hermine, O.; Bazarbachi, A.; Ratner, L.; Ramos, J.C.; Harrington, W., Jr.; O’Mahony, D.; Janik, J.E.; Bittencourt, A.L.; Taylor, G.P.; et al. Definition, prognostic factors, treatment, and response criteria of adult t-cell leukemia-lymphoma: A proposal from an international consensus meeting. J. Clin. Oncol. 2009, 27, 453–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gessain, A.; Cassar, O. Epidemiological aspects and world distribution of htlv-1 infection. Front. Microbiol. 2012, 3, 388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimoyama, M. Diagnostic criteria and classification of clinical subtypes of adult t- cell leukaemia-lymphoma. A report from the lymphoma study group (1984–87). Br. J. Haematol. 1991, 79, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Gessain, A.; Barin, F.; Vernant, J.C.; Gout, O.; Maurs, L.; Calender, A.; de The, G. Antibodies to human t-lymphotropic virus type-i in patients with tropical spastic paraparesis. Lancet 1985, 2, 407–410. [Google Scholar] [CrossRef]
- Osame, M.; Matsumoto, M.; Usuku, K.; Izumo, S.; Ijichi, N.; Amitani, H.; Tara, M.; Igata, A. Chronic progressive myelopathy associated with elevated antibodies to human t-lymphotropic virus type i and adult t-cell leukemialike cells. Ann. Neurol. 1987, 21, 117–122. [Google Scholar] [CrossRef]
- Einsiedel, L.; Pham, H.; Wilson, K.; Walley, R.; Turpin, J.; Bangham, C.; Gessain, A.; Woodman, R.J. Human t-lymphotropic virus type 1c subtype proviral loads, chronic lung disease and survival in a prospective cohort of indigenous australians. PLoS Negl. Trop. Dis. 2018, 12, e0006281. [Google Scholar] [CrossRef]
- Einsiedel, L.; Chiong, F.; Jersmann, H.; Taylor, G.P. Human t-cell leukaemia virus type 1 associated pulmonary disease: Clinical and pathological features of an under-recognised complication of htlv-1 infection. Retrovirology 2021, 18, 1. [Google Scholar] [CrossRef]
- Umeki, K.; Hisada, M.; Maloney, E.M.; Hanchard, B.; Okayama, A. Proviral loads and clonal expansion of htlv-1-infected cells following vertical transmission: A 10-year follow-up of children in jamaica. Intervirology 2009, 52, 115–122. [Google Scholar] [CrossRef] [Green Version]
- Giam, C.Z. Htlv-1 replication and adult t cell leukemia development. Recent Results Cancer Res. 2021, 217, 209–243. [Google Scholar]
- Matsuoka, M.; Mesnard, J.M. Htlv-1 bzip factor: The key viral gene for pathogenesis. Retrovirology 2020, 17, 2. [Google Scholar] [CrossRef]
- Harhaj, E.W.; Giam, C.Z. Nf-kappab signaling mechanisms in htlv-1-induced adult t-cell leukemia/lymphoma. FEBS J. 2018, 285, 3324–3336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majone, F.; Semmes, O.J.; Jeang, K.T. Induction of micronuclei by htlv-i tax: A cellular assay for function. Virology 1993, 193, 456–459. [Google Scholar] [CrossRef] [PubMed]
- Marriott, S.J.; Semmes, O.J. Impact of htlv-i tax on cell cycle progression and the cellular DNA damage repair response. Oncogene 2005, 24, 5986–5995. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, K.; Nagata, Y.; Kitanaka, A.; Shiraishi, Y.; Shimamura, T.; Yasunaga, J.; Totoki, Y.; Chiba, K.; Sato-Otsubo, A.; Nagae, G.; et al. Integrated molecular analysis of adult t cell leukemia/lymphoma. Nat. Genet. 2015, 47, 1304–1315. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Maeda, M.; Morikawa, S.; Taniguchi, Y.; Yasunaga, J.; Nosaka, K.; Tanaka, Y.; Matsuoka, M. Genetic and epigenetic inactivation of tax gene in adult t-cell leukemia cells. Int. J. Cancer 2004, 109, 559–567. [Google Scholar] [CrossRef]
- Giam, C.Z.; Semmes, O.J. Htlv-1 infection and adult t-cell leukemia/lymphoma-a tale of two proteins: Tax and hbz. Viruses 2016, 8, 161. [Google Scholar] [CrossRef]
- Matsuoka, M.; Green, P.L. The hbz gene, a key player in htlv-1 pathogenesis. Retrovirology 2009, 6, 71. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, M.; Shaffer, A.L., 3rd; Ceribelli, M.; Zhang, M.; Wright, G.; Huang, D.W.; Xiao, W.; Powell, J.; Petrus, M.N.; Yang, Y.; et al. Targeting the htlv-i-regulated batf3/irf4 transcriptional network in adult t cell leukemia/lymphoma. Cancer Cell 2018, 34, 286–297.e10. [Google Scholar] [CrossRef] [Green Version]
- Mahgoub, M.; Yasunaga, J.I.; Iwami, S.; Nakaoka, S.; Koizumi, Y.; Shimura, K.; Matsuoka, M. Sporadic on/off switching of htlv-1 tax expression is crucial to maintain the whole population of virus-induced leukemic cells. Proc. Natl. Acad. Sci. USA 2018, 115, E1269–E1278. [Google Scholar] [CrossRef] [Green Version]
- Miura, M.; Dey, S.; Ramanayake, S.; Singh, A.; Rueda, D.S.; Bangham, C.R.M. Kinetics of htlv-1 reactivation from latency quantified by single-molecule rna fish and stochastic modelling. PLoS Pathog. 2019, 15, e1008164. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Pasupala, N.; Zhi, H.; Dorjbal, B.; Hussain, I.; Shih, H.M.; Bhattacharyya, S.; Biswas, R.; Miljkovic, M.; Semmes, O.J.; et al. Nf-kappab-induced r-loop accumulation and DNA damage select for nucleotide excision repair deficiencies in adult t cell leukemia. Proc. Natl. Acad. Sci. USA 2021, 118, e2005568118. [Google Scholar] [CrossRef] [PubMed]
- Sollier, J.; Stork, C.T.; Garcia-Rubio, M.L.; Paulsen, R.D.; Aguilera, A.; Cimprich, K.A. Transcription-coupled nucleotide excision repair factors promote r-loop-induced genome instability. Mol. Cell 2014, 56, 777–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sollier, J.; Cimprich, K.A. Breaking bad: R-loops and genome integrity. Trends Cell Biol. 2015, 25, 514–522. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Muse, T.; Aguilera, A. R loops: From physiological to pathological roles. Cell 2019, 179, 604–618. [Google Scholar] [CrossRef] [PubMed]
- Wahba, L.; Amon, J.D.; Koshland, D.; Vuica-Ross, M. Rnase h and multiple rna biogenesis factors cooperate to prevent rna: DNA hybrids from generating genome instability. Mol. Cell 2011, 44, 978–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amon, J.D.; Koshland, D. Rnase h enables efficient repair of r-loop induced DNA damage. Elife 2016, 5, e20533. [Google Scholar] [CrossRef]
- Li, X.; Manley, J.L. Inactivation of the sr protein splicing factor asf/sf2 results in genomic instability. Cell 2005, 122, 365–378. [Google Scholar] [CrossRef] [Green Version]
- Gan, W.; Guan, Z.; Liu, J.; Gui, T.; Shen, K.; Manley, J.L.; Li, X. R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev. 2011, 25, 2041–2056. [Google Scholar] [CrossRef] [Green Version]
- Stork, C.T.; Bocek, M.; Crossley, M.P.; Sollier, J.; Sanz, L.A.; Chedin, F.; Swigut, T.; Cimprich, K.A. Co-transcriptional r-loops are the main cause of estrogen-induced DNA damage. Elife 2016, 5, e17548. [Google Scholar] [CrossRef]
- Kotsantis, P.; Silva, L.M.; Irmscher, S.; Jones, R.M.; Folkes, L.; Gromak, N.; Petermann, E. Increased global transcription activity as a mechanism of replication stress in cancer. Nat. Commun. 2016, 7, 13087. [Google Scholar] [CrossRef]
- Ho, Y.K.; Zhi, H.; Bowlin, T.; Dorjbal, B.; Philip, S.; Zahoor, M.A.; Shih, H.M.; Semmes, O.J.; Schaefer, B.; Glover, J.N.; et al. Htlv-1 tax stimulates ubiquitin e3 ligase, ring finger protein 8, to assemble lysine 63-linked polyubiquitin chains for tak1 and ikk activation. PLoS Pathog. 2015, 11, e1005102. [Google Scholar] [CrossRef] [PubMed]
- Shibata, Y.; Tokunaga, F.; Goto, E.; Komatsu, G.; Gohda, J.; Saeki, Y.; Tanaka, K.; Takahashi, H.; Sawasaki, T.; Inoue, S.; et al. Htlv-1 tax induces formation of the active macromolecular ikk complex by generating lys63- and met1-linked hybrid polyubiquitin chains. PLoS Pathog. 2017, 13, e1006162. [Google Scholar] [CrossRef] [PubMed]
- Uhlik, M.; Good, L.; Xiao, G.; Harhaj, E.W.; Zandi, E.; Karin, M.; Sun, S.C. Nf-kappab-inducing kinase and ikappab kinase participate in human t-cell leukemia virus i tax-mediated nf-kappab activation. J. Biol. Chem. 1998, 273, 21132–21136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhi, H.; Guo, X.; Ho, Y.K.; Pasupala, N.; Engstrom, H.A.A.; Semmes, O.J.; Giam, C.Z. Rnf8 dysregulation and down-regulation during htlv-1 infection promote genomic instability in adult t-cell leukemia. PLoS Pathog. 2020, 16, e1008618. [Google Scholar] [CrossRef]
- Semmes, O.J.; Jeang, K.T. Localization of human t-cell leukemia virus type 1 tax to subnuclear compartments that overlap with interchromatin speckles. J. Virol. 1996, 70, 6347–6357. [Google Scholar] [CrossRef] [Green Version]
- Durkin, S.S.; Guo, X.; Fryrear, K.A.; Mihaylova, V.T.; Gupta, S.K.; Belgnaoui, S.M.; Haoudi, A.; Kupfer, G.M.; Semmes, O.J. Htlv-1 tax oncoprotein subverts the cellular DNA damage response via binding to DNA-dependent protein kinase. J. Biol. Chem. 2008, 283, 36311–36320. [Google Scholar] [CrossRef] [Green Version]
- Belgnaoui, S.M.; Fryrear, K.A.; Nyalwidhe, J.O.; Guo, X.; Semmes, O.J. The viral oncoprotein tax sequesters DNA damage response factors by tethering mdc1 to chromatin. J. Biol. Chem. 2010, 285, 32897–32905. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 years of nf-kappab: A blossoming of relevance to human pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef] [Green Version]
- Lohr, J.G.; Stojanov, P.; Lawrence, M.S.; Auclair, D.; Chapuy, B.; Sougnez, C.; Cruz-Gordillo, P.; Knoechel, B.; Asmann, Y.W.; Slager, S.L.; et al. Discovery and prioritization of somatic mutations in diffuse large b-cell lymphoma (dlbcl) by whole-exome sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, 3879–3884. [Google Scholar] [CrossRef] [Green Version]
- Morin, R.D.; Mungall, K.; Pleasance, E.; Mungall, A.J.; Goya, R.; Huff, R.D.; Scott, D.W.; Ding, J.; Roth, A.; Chiu, R.; et al. Mutational and structural analysis of diffuse large b-cell lymphoma using whole-genome sequencing. Blood 2013, 122, 1256–1265. [Google Scholar] [CrossRef] [Green Version]
- Ngo, V.N.; Young, R.M.; Schmitz, R.; Jhavar, S.; Xiao, W.; Lim, K.H.; Kohlhammer, H.; Xu, W.; Yang, Y.; Zhao, H.; et al. Oncogenically active myd88 mutations in human lymphoma. Nature 2011, 470, 115–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasqualucci, L.; Trifonov, V.; Fabbri, G.; Ma, J.; Rossi, D.; Chiarenza, A.; Wells, V.A.; Grunn, A.; Messina, M.; Elliot, O.; et al. Analysis of the coding genome of diffuse large b-cell lymphoma. Nat. Genet. 2011, 43, 830–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, M.A.; Lawrence, M.S.; Keats, J.J.; Cibulskis, K.; Sougnez, C.; Schinzel, A.C.; Harview, C.L.; Brunet, J.P.; Ahmann, G.J.; Adli, M.; et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011, 471, 467–472. [Google Scholar] [CrossRef]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabbri, G.; Dalla-Favera, R. The molecular pathogenesis of chronic lymphocytic leukaemia. Nat. Rev. Cancer 2016, 16, 145–162. [Google Scholar] [CrossRef]
- Matsuoka, M.; Jeang, K.T. Human t-cell leukaemia virus type 1 (htlv-1) infectivity and cellular transformation. Nat. Rev. Cancer 2007, 7, 270–280. [Google Scholar] [CrossRef]
- McGuire, K.L.; Curtiss, V.E.; Larson, E.L.; Haseltine, W.A. Influence of human t-cell leukemia virus type i tax and rex on interleukin-2 gene expression. J. Virol. 1993, 67, 1590–1599. [Google Scholar] [CrossRef] [Green Version]
- Siekevitz, M.; Feinberg, M.B.; Holbrook, N.; Wong Staal, F.; Greene, W.C. Activation of interleukin 2 and interleukin 2 receptor (tac) promoter expression by the trans-activator (tat) gene product of human t-cell leukemia virus, type i. Proc. Natl. Acad. Sci. USA 1987, 84, 5389–5393. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, M.; Shibuya, H.; Harada, H.; Hatakeyama, M.; Seiki, M.; Fujita, T.; Inoue, J.; Yoshida, M.; Taniguchi, T. Evidence for aberrant activation of the interleukin-2 autocrine loop by htlv-1-encoded p40x and t3/ti complex triggering. Cell 1987, 48, 343–350. [Google Scholar] [CrossRef]
- Kuo, Y.L.; Giam, C.Z. Activation of the anaphase promoting complex by htlv-1 tax leads to senescence. EMBO J. 2006, 25, 1741–1752. [Google Scholar] [CrossRef] [Green Version]
- Zhi, H.; Zahoor, M.A.; Shudofsky, A.M.; Giam, C.Z. Kshv vcyclin counters the senescence/g1 arrest response triggered by nf-kappab hyperactivation. Oncogene 2014, 34, 496–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Zhi, H.; Liu, M.; Kuo, Y.L.; Giam, C.Z. Induction of p21(cip1/waf1) expression by human t-lymphotropic virus type 1 tax requires transcriptional activation and mrna stabilization. Retrovirology 2009, 6, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhi, H.; Yang, L.; Kuo, Y.L.; Ho, Y.K.; Shih, H.M.; Giam, C.Z. Nf-kappab hyper-activation by htlv-1 tax induces cellular senescence, but can be alleviated by the viral anti-sense protein hbz. PLoS Pathog. 2011, 7, e1002025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, Y.K.; Zhi, H.; Debiaso, D.; Philip, S.; Shih, H.M.; Giam, C.Z. Htlv-1 tax-induced rapid senescence is driven by the transcriptional activity of nf-kappab and depends on chronically activated ikkalpha and p65/rela. J. Virol. 2012, 86, 9474–9483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Yang, L.; Zhang, L.; Liu, B.; Merling, R.; Xia, Z.; Giam, C.Z. Human t-cell leukemia virus type 1 infection leads to arrest in the g1 phase of the cell cycle. J. Virol. 2008, 82, 8442–8455. [Google Scholar] [CrossRef] [Green Version]
- Philip, S.; Zahoor, M.A.; Zhi, H.; Ho, Y.K.; Giam, C.Z. Regulation of human t-lymphotropic virus type i latency and reactivation by hbz and rex. PLoS Pathog. 2014, 10, e1004040. [Google Scholar] [CrossRef]
- Zahoor, M.A.; Philip, S.; Zhi, H.; Giam, C.Z. Nf-kappab inhibition facilitates the establishment of cell lines that chronically produce human t-lymphotropic virus type 1 viral particles. J. Virol. 2014, 88, 3496–3504. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Kotomura, N.; Ho, Y.K.; Zhi, H.; Bixler, S.; Schell, M.J.; Giam, C.Z. Complex cell cycle abnormalities caused by human t-lymphotropic virus type 1 tax. J. Virol. 2011, 85, 3001–3009. [Google Scholar] [CrossRef] [Green Version]
- Shudofsky, A.M.D.; Giam, C.Z. Cells of adult t-cell leukemia evade htlv-1 tax/nf-kappab hyperactivation-induced senescence. Blood Adv. 2019, 3, 564–569. [Google Scholar] [CrossRef] [Green Version]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef]
- de Boer, J.; Hoeijmakers, J.H. Nucleotide excision repair and human syndromes. Carcinogenesis 2000, 21, 453–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costantino, L.; Koshland, D. The yin and yang of r-loop biology. Curr. Opin. Cell Biol. 2015, 34, 39–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skourti-Stathaki, K.; Proudfoot, N.J. A double-edged sword: R loops as threats to genome integrity and powerful regulators of gene expression. Genes Dev. 2014, 28, 1384–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos-Pereira, J.M.; Aguilera, A. R loops: New modulators of genome dynamics and function. Nat. Rev. Genet. 2015, 16, 583–597. [Google Scholar] [CrossRef]
- Richard, P.; Manley, J.L. R loops and links to human disease. J. Mol. Biol. 2017, 429, 3168–3180. [Google Scholar] [CrossRef] [Green Version]
- Laman, H.; Coverley, D.; Krude, T.; Laskey, R.; Jones, N. Viral cyclin-cyclin-dependent kinase 6 complexes initiate nuclear DNA replication. Mol. Cell Biol. 2001, 21, 624–635. [Google Scholar] [CrossRef] [Green Version]
- Verschuren, E.W.; Jones, N.; Evan, G.I. The cell cycle and how it is steered by kaposi’s sarcoma-associated herpesvirus cyclin. J. Gen. Virol. 2004, 85, 1347–1361. [Google Scholar] [CrossRef]
- Crossley, M.P.; Bocek, M.; Cimprich, K.A. R-loops as cellular regulators and genomic threats. Mol. Cell 2019, 73, 398–411. [Google Scholar] [CrossRef] [Green Version]
- Asquith, B.; Hanon, E.; Taylor, G.P.; Bangham, C.R.M. Is human t-cell lymphotropic virus type i really silent? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2000, 355, 1013–1019. [Google Scholar] [CrossRef]
- Okayama, A.; Stuver, S.; Matsuoka, M.; Ishizaki, J.; Tanaka, G.; Kubuki, Y.; Mueller, N.; Hsieh, C.C.; Tachibana, N.; Tsubouchi, H. Role of htlv-1 proviral DNA load and clonality in the development of adult t-cell leukemia/lymphoma in asymptomatic carriers. Int. J. Cancer 2004, 110, 621–625. [Google Scholar] [CrossRef]
- Cereseto, A.; Diella, F.; Mulloy, J.C.; Cara, A.; Michieli, P.; Grassmann, R.; Franchini, G.; Klotman, M.E. P53 functional impairment and high p21waf1/cip1 expression in human t-cell lymphotropic/leukemia virus type i-transformed t cells. Blood 1996, 88, 1551–1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pise-Masison, C.A.; Mahieux, R.; Jiang, H.; Ashcroft, M.; Radonovich, M.; Duvall, J.; Guillerm, C.; Brady, J.N. Inactivation of p53 by human t-cell lymphotropic virus type 1 tax requires activation of the nf-kappab pathway and is dependent on p53 phosphorylation. Mol. Cell Biol. 2000, 20, 3377–3386. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giam, C.-Z.; Pasupala, N. NF-κB-Induced R-Loops and Genomic Instability in HTLV-1-Infected and Adult T-Cell Leukemia Cells. Viruses 2022, 14, 877. https://doi.org/10.3390/v14050877
Giam C-Z, Pasupala N. NF-κB-Induced R-Loops and Genomic Instability in HTLV-1-Infected and Adult T-Cell Leukemia Cells. Viruses. 2022; 14(5):877. https://doi.org/10.3390/v14050877
Chicago/Turabian StyleGiam, Chou-Zen, and Nagesh Pasupala. 2022. "NF-κB-Induced R-Loops and Genomic Instability in HTLV-1-Infected and Adult T-Cell Leukemia Cells" Viruses 14, no. 5: 877. https://doi.org/10.3390/v14050877
APA StyleGiam, C.-Z., & Pasupala, N. (2022). NF-κB-Induced R-Loops and Genomic Instability in HTLV-1-Infected and Adult T-Cell Leukemia Cells. Viruses, 14(5), 877. https://doi.org/10.3390/v14050877